一种噻螨酮及其制备方法与流程
本发明涉及杀虫剂领域,具体涉及一种噻螨酮及其制备方法。
背景技术:
国家对农药的发展越来越重视,在环保方面,降低农药对社会和环境的风险成为重中之重。随着种植结构的变化,农药三大系列之一的杀虫剂(杀螨剂)近年来,销量获得了快速增长,世界各国也加大了农药技术开发力度,新技术、新产品不断涌现。
噻螨酮,化学名称为:(4RS,5RS)-5-(4-氯苯基)-N-环己基-4-甲基-2-氧代-1,3-噻唑烷-3-羧酰胺。其化学结构式为:
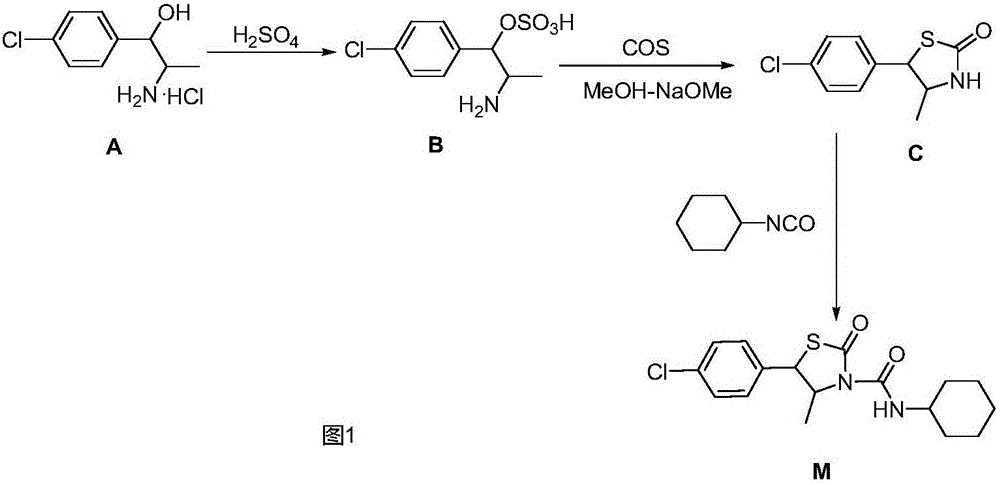
该产品自1985年日本曹达公司开发,其合成路线如图1所示:以赤-2-氨基-1-(对氯苯基)丙醇盐酸盐(A)为起始原料,经硫酸缩合脱水而生成赤-[2-氨基-1-(对氯苯基)]丙基硫酸酯(赤-硫酸酯)(B),然后与氧硫化碳闭环生成取代的反式噻唑烷酮(C),后者再与异氰酸环己酯加成而得到最终产物噻螨酮(M)。该合成工艺所用的起始原料需经多步化学反应合成;其关键性中间体反式噻唑烷酮(C)由中间体赤-硫酸酯(B)与氧硫化碳进行关环反应制备,由于氧硫化碳需通过一氧化碳同硫磺蒸汽作用而得到,一氧化碳为剧毒气体,对工艺设备和操作均有严格要求,而且污染环境。因此,该合成工艺存在有工艺流程复杂、合成难度大、操作不安全、污染环境、成本高等缺点。
随后有人报道对此工艺进行改进,制备方法如图2所示:采用二硫化碳进行环化,环化后再用双氧水进行氧化得到反式噻唑烷酮(C),后者再与异氰酸环己酯加成而得到最终产物噻螨酮(M)。但改进后依然需要对原料A预先硫酸酯化,这一步反应往往会造成大量的酸性废水。
另外有报道,制备方法如图3所示:以赤-2-氨基-1-(对氯苯基)丙醇盐酸盐(A)为原料,在碱存在下与二硫化碳及苄氯反应生成化合物(D),反应产物D在二氯甲烷中与氯化亚砜一起进行化合反应生成(E),然后,将生成物E在盐酸水溶液中加热进行回流开环成酮反应生成(F),再经环合得取代的反式噻唑烷酮(C),最后再与异氰酸环己酯加成而得到最终产物噻螨酮(M)。此路线反应步骤长、操作繁琐、污染较大、收率也低,不适合工业化生产。
因此,需要一种噻螨酮的绿色化生产工艺路线,适合工业化生产,能够简化工艺流程,提高反应收率,降低合成难度,减少副产物的形成,降低制备过程中的环境污染问题,并且降低生产成本。
技术实现要素:
本发明针对上述问题,提供一种噻螨酮及其制备方法,适合工业化生产,能够简化工艺流程,提高反应收率,降低合成难度,减少副产物的形成,降低制备过程中的环境污染问题,并且降低生产成本。
本发明解决上述问题所采用的技术方案是:一种噻螨酮的制备方法,以对氯苯丙酮为起始原料,和亚硝酸正丁酯以70:47的质量比反应生成酮肟物,然后进行氢化还原为2-氨基-1-对氯苯基丙醇盐酸盐。
进一步地,2-氨基-1-对氯苯基丙醇盐酸盐直接与二硫化碳反应合成反式-5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮,再经双氧水氧化生成5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮,5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮再同异氰酸环己酯进行加成反应而得到最终产物噻螨酮。
进一步地,酮肟物的制备过程具体为:向反应釜中投入350份对氯苯丙酮、700份石油醚,降温到10℃~20℃,再加入235份亚硝酸正丁酯,搅拌5h~10h,再降温至0℃,保温搅拌0.5h~1.5h,经放料、过滤、石油醚洗涤,烘干得到固体酮肟物。
进一步地,2-氨基-1-对氯苯基丙醇盐酸盐的制备过程具体为:在反应釜中投入180份制得的酮肟物、480份甲醇,加入催化剂A 1份~2份,溶解0.5h,氮气置换2次~4次,再氢气置换2次~4次,升温至35℃~40℃,通入氢气,控制氢气压力为0.03Mpa~0.08Mpa,直到加不进氢气为止,通氢气完毕,趁热过滤除去催化剂A,得到醇胺物的甲醇溶液,用盐酸调PH值到6~6.5,减压脱去溶剂得到醇胺物粗品,冷却至40℃~50℃,加入500份乙醇,升温回流1h~3h,冷却到0℃~5℃后,保温1h~3h,所得产物经离心、烘干,得2-氨基-1-对氯苯基丙醇盐酸盐。
更进一步地,催化剂A包括:铂、钯、镍中的任一种。
更进一步地,噻螨酮的制备方法,包括以下步骤:
步骤S1,向反应釜中投入350份对氯苯丙酮、700份石油醚,降温到10℃~20℃,再加入235份亚硝酸正丁酯,搅拌5h~10h,再降温至0℃,保温搅拌0.5h~1.5h,经放料、过滤、石油醚洗涤,烘干得到固体酮肟物;
步骤S2,在反应釜中投入180份制得的酮肟物、480份甲醇,加入催化剂A1份~2份,溶解0.5h,氮气置换2次~4次,再氢气置换2次~4次,升温至35℃~40℃,通入氢气,控制氢气压力为0.03Mpa~0.08Mpa,直到加不进氢气为止,通氢气完毕,趁热过滤除去催化剂A,得到醇胺物的甲醇溶液,用盐酸调PH值到6~6.5,减压脱去溶剂得到醇胺物粗品,冷却至40℃~50℃,加入500份乙醇,升温回流1h~3h,冷却到0℃~5℃后,保温1h~3h,所得产物经离心、烘干,得2-氨基-1-对氯苯基丙醇盐酸盐;
步骤S3,在反应釜中投入2-氨基-1-对氯苯基丙醇盐酸盐180份,8%氢氧化钾溶液700份、催化剂B1份~3份,搅拌均匀后,再加入二硫化碳77份,然后在80℃~100℃搅拌反应12h~14h,保温结束后降温到常温,然后反应釜中抽入500份石油醚,搅拌0.5h~1.5h,静置0.5h~2h,分层,有机层去脱溶,脱溶结束后,加入甲醇500份升温回流2h~4h,降温到0℃~5℃析出晶体后,再保温1h~3h,离心烘干得到5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮;
步骤S4,在反应釜中抽入甲醇800份,投入5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮190份,催化剂C 2份~4份,开启搅拌,然后在25℃加入300份的28%甲醇钠,加毕后,搅拌半小时,然后在25℃以下滴加160份30%双氧水,滴加完毕,在15℃~25℃保温4h~8h,保温结束后,转入脱溶釜,减压脱去甲醇,降温到室温,抽入甲苯700份,水500份,搅拌半小时,静置半小时,分去水层,减压脱去甲苯,降温到室温,加入500份甲醇,升温回流1h~3h,降温到0℃~5℃保温2h~4h,抽滤得到5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮;
步骤S5,在反应釜中,加入700份甲苯、200份5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮和10份催化剂D,调节反应釜温度为35℃~40℃,开始滴加125份异氰酸环已酯,时间控制在1.5h~2.5h,滴完后,保温反应4h~8h,取样中控,合格后,加入400份水,控制温度为35℃~40℃,继续搅拌1h。然后将反应混合物抽滤,滤液分去水层,油层用400份水洗一次,分去水层,抽入脱溶釜减压蒸出甲苯,冷却到50℃~60℃,加入520份乙醇,升温回流2h~4h,冷却到5℃~10℃保温1h~3h,产物经离心、烘干,得到噻螨酮原药。
更进一步地,步骤S3中,催化剂B为:甲醇钠或氢氧化钠。
更进一步地,步骤S4中,催化剂C包括:过氧化钠或甲醇钠。
更进一步地,步骤S5中,催化剂D包括:二氯甲烷、四氢呋喃、三乙胺、二月桂酸二丁基锡中的任一种。
本发明的另一发明目的,在于提供一种噻螨酮,根据上述制备方法制备而成。
本发明的优点是:
1.本发明用绿色化合成工艺开发了噻螨酮农药,该农药具有广谱、低毒、高效、低剂量等优点,有强烈的杀卵、幼、若螨作用,和其他杀螨剂相比,杀卵的效果更为显著,对植物表皮具有良好的渗透性,每亩单位用量较低仅需有效成分3g~5g,且药效稳定,持效期长,不受温度影响,广泛用于柑橘、苹果、山楂、棉花、蔬菜等农作物上防止害螨,对人体、环境较为安全,是更新换代的理想产品,具有良好的市场发展前景;
2.本发明在其合成反应的全过程中,所用的原材料绝大部分转化到最终产物,使用溶剂予以回收循环利用,提高了原子利用率与原子经济性,减少环境污染;
3.本发明采用一定压力条件下的二硫化碳成环技术,替代先硫酸酯化再成环的技术和有毒有污染的硫氧化碳成环工艺,大大降低了酸性废水的排放;
4.本发明采用高效选择性催化剂,以提高反应收率、减少副产物的形成并降低环境污染;
5.本发明采用石油醚、乙醇等低毒溶剂替代二甲基甲酰胺、二甲亚砜高毒溶剂,以最大限度地降低环境污染。
附图说明
构成本说明书的一部分、用于进一步理解本发明的附图示出了本发明的实施方案,并与说明书一起用来说明本发明的制备流程。在附图中:
图1是现有技术中日本曹达公司制备噻螨酮的合成技术路线;
图2是现有技术中一种制备噻螨酮的合成技术路线;
图3是现有技术中另一种制备噻螨酮的合成技术路线;
图4是本发明中噻螨酮原药的制备工艺流程图;
图5是本发明中噻螨酮原药的红外光谱谱图;
图6是本发明中噻螨酮原药的紫外吸收光谱图;
图7是本发明中噻螨酮原药的核磁共振吸收图谱;
图8是本发明中噻螨酮原药的有效成分质谱图。
具体实施方式
以下结合附图对本发明的实施例进行详细说明,但是本发明可以由权利要求限定和覆盖的多种不同方式实施。
实施例1
一种噻螨酮的制备方法,包括以下步骤:
步骤S1,酮肟化反应
反应方程式为:
原料中:对氯苯丙酮,纯度99%;亚硝酸正丁酯,纯度99%;石油醚,温度90℃~120℃;
操作:向3000升反应釜中投入350Kg对氯苯丙酮、700Kg石油醚,降温到10℃,再加入235Kg亚硝酸正丁酯,搅拌5h,再降温至0℃,保温搅拌0.5h,经放料、过滤、石油醚洗涤,烘干得到固体酮肟物;
步骤S2,醇胺化反应
反应方程式为:
原料中:酮肟物,纯度99%;甲醇,纯度99.5%;盐酸,纯度30%;乙醇,纯度99.5%;
操作:在2000升反应釜中投入180Kg制得的酮肟物、480Kg甲醇,加入催化剂铂1Kg,溶解0.5h,氮气置换2次,再氢气置换2次,升温至35℃,通入氢气,控制氢气压力为0.03Mpa,直到加不进氢气为止,通氢气完毕,趁热过滤除去催化剂铂,得到醇胺物的甲醇溶液,用盐酸调PH值到6,减压脱去溶剂得到醇胺物粗品,冷却至40℃,加入500Kg乙醇,升温回流1h,冷却到0℃后,保温1h,所得产物经离心、烘干,得2-氨基-1-对氯苯基丙醇盐酸盐;
步骤S3,环合反应
反应方程式为:
原料中:氢氧化钾,质量百分数8%;
操作:在2000升反应釜中投入2-氨基-1-对氯苯基丙醇盐酸盐180Kg,8%氢氧化钾溶液700Kg、甲醇钠1Kg,搅拌均匀后,再加入二硫化碳77Kg份,然后在80℃搅拌反应12h,保温结束后降温到常温,然后反应釜中抽入500Kg石油醚,搅拌0.5h,静置0.5h,分层,有机层去脱溶,脱溶结束后,加入甲醇500Kg升温回流2h,降温到0℃析出晶体后,再保温1h,离心烘干得到5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮;
步骤S4,氧化反应
反应方程式为:
原料中:甲醇钠的质量分数为28%;双氧水的质量分数为30%;
操作:在3000升反应釜中抽入甲醇800Kg,投入5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮190Kg,过氧化钠2Kg,开启搅拌,然后在25℃加入300Kg的甲醇钠,加毕后,搅拌半小时,然后在25℃以下滴加160Kg双氧水,滴加完毕,在15℃保温4h,保温结束后,转入脱溶釜,减压脱去甲醇,降温到室温,抽入甲苯700Kg,水500Kg,搅拌半小时,静置半小时,分去水层,减压脱去甲苯,降温到室温,加入500Kg甲醇,升温回流1h,降温到0℃保温2h,抽滤得到5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮;
步骤S5,加成反应
反应方程式为:
原料中:5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮,纯度97%;异氰酸环已酯,纯度为:99%;
操作:在2000升反应釜中,加入700Kg甲苯、200Kg 5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮和10Kg二氯甲烷,调节反应釜温度为35℃,开始滴加125Kg异氰酸环已酯,时间控制在1.5h,滴完后,保温反应4h,取样中控,合格后,加入400Kg水,控制温度为35℃,继续搅拌1h。然后将反应混合物抽滤,滤液分去水层,油层用400Kg水洗一次,分去水层,抽入脱溶釜减压蒸出甲苯,冷却到50℃,加入520Kg乙醇,升温回流2h,冷却到5℃保温1h,产物经离心、烘干,得到噻螨酮原药。
实施例2
一种噻螨酮的制备方法,包括以下步骤:
步骤S1,向3000升反应釜中投入174Kg对氯苯丙酮、350Kg石油醚,降温到20℃,再加入117.5Kg亚硝酸正丁酯,搅拌10h,再降温至0℃,保温搅拌1.5h,经放料、过滤、石油醚洗涤,烘干得到固体酮肟物;
步骤S2,在2000升反应釜中投入90Kg制得的酮肟物、240Kg甲醇,加入催化剂钯1Kg,溶解0.5h,氮气置换4次,再氢气置换4次,升温至40℃,通入氢气,控制氢气压力为0.08Mpa,直到加不进氢气为止,通氢气完毕,趁热过滤除去催化剂钯,得到醇胺物的甲醇溶液,用盐酸调PH值到6.5,减压脱去溶剂得到醇胺物粗品,冷却至50℃,加入250Kg乙醇,升温回流3h,冷却到5℃后,保温3h,所得产物经离心、烘干,得2-氨基-1-对氯苯基丙醇盐酸盐;
步骤S3,在2000升反应釜中投入2-氨基-1-对氯苯基丙醇盐酸盐90Kg,8%氢氧化钾溶液350Kg、氢氧化钠1.5Kg,搅拌均匀后,再加入二硫化碳38.5Kg份,然后在100℃搅拌反应14h,保温结束后降温到常温,然后反应釜中抽入250Kg石油醚,搅拌1.5h,静置2h,分层,有机层去脱溶,脱溶结束后,加入甲醇250Kg升温回流4h,降温到5℃析出晶体后,再保温3h,离心烘干得到5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮;
步骤S4,在3000升反应釜中抽入甲醇400Kg,投入5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮95Kg,甲醇钠2Kg,开启搅拌,然后在25℃加入150Kg的甲醇钠,加毕后,搅拌半小时,然后在25℃以下滴加80Kg双氧水,滴加完毕,在25℃保温8h,保温结束后,转入脱溶釜,减压脱去甲醇,降温到室温,抽入甲苯350Kg,水250Kg,搅拌半小时,静置半小时,分去水层,减压脱去甲苯,降温到室温,加入250Kg甲醇,升温回流3h,降温到5℃保温4h,抽滤得到5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮;
步骤S5,在2000升反应釜中,加入350Kg甲苯、100Kg 5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮和5Kg四氢呋喃,调节反应釜温度为40℃,开始滴加62.5Kg异氰酸环已酯,时间控制在2.5h,滴完后,保温反应8h,取样中控,合格后,加入200Kg水,控制温度为40℃,继续搅拌1h。然后将反应混合物抽滤,滤液分去水层,油层用200Kg水洗一次,分去水层,抽入脱溶釜减压蒸出甲苯,冷却到60℃,加入260Kg乙醇,升温回流4h,冷却到10℃保温3h,产物经离心、烘干,得到噻螨酮原药。
实施例3
一种噻螨酮的制备方法,包括以下步骤:
步骤S1,向3000升反应釜中投入420Kg对氯苯丙酮、840Kg石油醚,降温到15℃,再加入282Kg亚硝酸正丁酯,搅拌7.5h,再降温至0℃,保温搅拌1h,经放料、过滤、石油醚洗涤,烘干得到固体酮肟物;
步骤S2,在2000升反应釜中投入216Kg制得的酮肟物、576Kg甲醇,加入催化剂镍1.8Kg,溶解0.5h,氮气置换3次,再氢气置换3次,升温至37℃,通入氢气,控制氢气压力为0.05Mpa,直到加不进氢气为止,通氢气完毕,趁热过滤除去催化剂镍,得到醇胺物的甲醇溶液,用盐酸调PH值到6.3,减压脱去溶剂得到醇胺物粗品,冷却至45℃,加入600Kg乙醇,升温回流2h,冷却到2.5℃后,保温2h,所得产物经离心、烘干,得2-氨基-1-对氯苯基丙醇盐酸盐;
步骤S3,在2000升反应釜中投入2-氨基-1-对氯苯基丙醇盐酸盐216Kg,8%氢氧化钾溶液840Kg、甲醇钠2.4Kg,搅拌均匀后,再加入二硫化碳92.4Kg,然后在90℃搅拌反应13h,保温结束后降温到常温,然后反应釜中抽入600Kg石油醚,搅拌1h,静置1.2h,分层,有机层去脱溶,脱溶结束后,加入甲醇600Kg升温回流3h,降温到2.5℃析出晶体后,再保温2h,离心烘干得到5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮;
步骤S4,在3000升反应釜中抽入甲醇960Kg,投入5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮228Kg,过氧化钠3.6Kg,开启搅拌,然后在25℃加入360Kg的甲醇钠,加毕后,搅拌半小时,然后在25℃以下滴加192Kg双氧水,滴加完毕,在20℃保温6h,保温结束后,转入脱溶釜,减压脱去甲醇,降温到室温,抽入甲苯840Kg,水600Kg,搅拌半小时,静置半小时,分去水层,减压脱去甲苯,降温到室温,加入600Kg甲醇,升温回流2h,降温到2.5℃保温3h,抽滤得到5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮;
步骤S5,在2000升反应釜中,加入840Kg甲苯、240Kg 5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮和12Kg三乙胺,调节反应釜温度为37℃,开始滴加150Kg异氰酸环已酯,时间控制在2h,滴完后,保温反应6h,取样中控,合格后,加入480Kg水,控制温度为37℃,继续搅拌1h。然后将反应混合物抽滤,滤液分去水层,油层用480Kg水洗一次,分去水层,抽入脱溶釜减压蒸出甲苯,冷却到55℃,加入624Kg乙醇,升温回流3h,冷却到7.5℃保温2h,产物经离心、烘干,得到噻螨酮原药。
实施例4
一种噻螨酮的制备方法,包括以下步骤:
步骤S1,向3000升反应釜中投入350Kg对氯苯丙酮、700Kg石油醚,降温到12℃,再加入235Kg亚硝酸正丁酯,搅拌8h,再降温至0℃,保温搅拌1.2h,经放料、过滤、石油醚洗涤,烘干得到固体酮肟物;
步骤S2,在2000升反应釜中投入180Kg制得的酮肟物、480Kg甲醇,加入催化剂镍1.8Kg,溶解0.5h,氮气置换2次,再氢气置换4次,升温至39℃,通入氢气,控制氢气压力为0.06Mpa,直到加不进氢气为止,通氢气完毕,趁热过滤除去催化剂镍,得到醇胺物的甲醇溶液,用盐酸调PH值到6.2,减压脱去溶剂得到醇胺物粗品,冷却至42℃,加入500Kg乙醇,升温回流2.5h,冷却到4℃后,保温1.5h,所得产物经离心、烘干,得2-氨基-1-对氯苯基丙醇盐酸盐;
步骤S3,在2000升反应釜中投入2-氨基-1-对氯苯基丙醇盐酸盐180Kg,8%氢氧化钾溶液700Kg、氢氧化钠1.5Kg,搅拌均匀后,再加入二硫化碳77Kg,然后在85℃搅拌反应12.5h,保温结束后降温到常温,然后反应釜中抽入500Kg石油醚,搅拌1.2h,静置1.5h,分层,有机层去脱溶,脱溶结束后,加入甲醇500Kg升温回流2.5h,降温到1℃析出晶体后,再保温1.5h,离心烘干得到5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮;
步骤S4,在3000升反应釜中抽入甲醇800Kg,投入5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮190Kg,甲醇钠3.5Kg,开启搅拌,然后在25℃加入300Kg的甲醇钠,加毕后,搅拌半小时,然后在25℃以下滴加160Kg双氧水,滴加完毕,在18℃保温5h,保温结束后,转入脱溶釜,减压脱去甲醇,降温到室温,抽入甲苯700Kg,水500Kg,搅拌半小时,静置半小时,分去水层,减压脱去甲苯,降温到室温,加入500Kg甲醇,升温回流1.5h,降温到1℃保温2.5h,抽滤得到5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮;
步骤S5,在2000升反应釜中,加入700Kg甲苯、200Kg 5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮和10Kg二月桂酸二丁基锡,调节反应釜温度为38℃,开始滴加125Kg异氰酸环已酯,时间控制在1.8h,滴完后,保温反应5h,取样中控,合格后,加入400Kg水,控制温度为37℃,继续搅拌1h。然后将反应混合物抽滤,滤液分去水层,油层用400Kg水洗一次,分去水层,抽入脱溶釜减压蒸出甲苯,冷却到52℃,加入520Kg乙醇,升温回流2.5h,冷却到6℃保温2.5h,产物经离心、烘干,得到噻螨酮原药。
实验例1
实施例1~4所制的酮肟物的试产数据如表1所示。
表1酮肟物试产数据
实施例1~4所制的2-氨基-1-对氯苯基丙醇盐酸盐(β-醇胺物)的试产数据如表2所示。
表2 2-氨基-1-对氯苯基丙醇盐酸盐(β-醇胺物)试产数据
实施例1~4所制的5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮的试产数据如表3所示。
表3 5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮试产数据
实施例1~4所制的5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮的试产数据如表4所示。
表4 5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮试产数据
实施例1~4所制的噻螨酮原药的试产数据如表5所示。
表5噻螨酮原药试产数据
由上述试验结果可知:酮肟化反应的平均收率为96.8%,所得酮肟物的平均含量≥99%;醇胺化反应的平均收率为95.4%,所得醇胺物的平均含量为≥99%;环合反应的平均收率为93.6%,所得环合物的平均含量≥96%;氧化反应的平均收率为90.8%,所得噻唑烷酮的平均含量≥97%;加成反应平均收率为95.8%,所得最终产物噻螨酮原药的平均含量为98.8%。
实验例2
对实施例1~4所制的噻螨酮原药进行分析及表征。
(1)红外光谱法:采用溴化钾压片法,压下制成溴化钾-农药样本薄片,以纯的溴化钾薄片为对照,在红外光谱仪上扫描样本即得红外光谱谱图,扫描范围为4000cm-400cm。
结果表明,如图5所示,实施例1~4所制的噻螨酮原药的有效成份与噻螨酮具有相同的红外光谱吸收特征。
(2)紫外光谱法:分别取适量待测样品用乙腈溶解,以乙腈为空白对照,按照仪器操作规程测定待测样品的紫外吸收。
如图6所示,实施例1~4所制的噻螨酮原药的紫外吸收光谱图之间无明显差异,皆与噻螨酮标样的紫外吸收光谱图十分吻合。从紫外吸收图谱可见:噻螨酮原药的紫外光谱与噻螨酮标准品的谱图相同,在约196nm和223nm附近有较强吸收。
结果表明,实施例1~4所制的噻螨酮原药的有效成份与噻螨酮具有相同的紫外光谱吸收特征。
(3)核磁共振法:将待测样品各约10mg,分别放入核磁管中,加入适量d-DMSO,盖紧管盖,振荡使样本溶解。按照仪器操作规程在核磁共振仪上测定。
如图7所示,实施例1~4所制的噻螨酮原药与噻螨酮标样的核磁共振吸收图谱之间无明显差异,各H原子的化学位移和裂分情况也相同。
结果表明,实施例1~4所制的噻螨酮原药的有效成份与噻螨酮具有相同的核磁共振波谱特征。
(4)质谱法:以液相色谱为分离工具,以MSD为检测器,在选定的色谱及离子化条件下,对噻螨酮原药中的主要组分进行分离。通过分析各实施例制备的噻螨酮原药中有效成分质谱图,结合原药生产工艺,对噻螨酮原药中有效成分进行分离与定性鉴定。
结果显示,如图8所示,实施例1~4所制的噻螨酮原药有效成分和标样的MS定性分析谱图之间无明显差异。根据噻螨酮原药的合成工艺和质谱数据给出的信息,推断出实施例1~4所制的噻螨酮原药有效成分是噻螨酮。
实验例3
对实施例1~4所制的噻螨酮原药进行全组分分析鉴定,鉴定结果如表6所示。
表6噻螨酮原药全组分分析鉴定结果
以上仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!