一种八臂杂臂星形聚合物及其制备方法与流程
本发明属于高分子合成领域,具体涉及一种八臂杂臂星形聚合物的制备方法。
背景技术:
星形聚合物一般是指由多个线型支链连接于同一中心核上所构成的一类支化聚合物。组成星形聚合物的线型支链称为星形聚合物的“臂”,而中心的多官能基团称为星形聚合物的“核”。根据“臂”的化学组成以及“核”结构的不同,可将星形聚合物大致分为同臂星形聚合物、杂臂星形聚合物以及树枝状星形聚合物。与线型聚合物相比,这类具有多臂拓扑结构的聚合物具有较小的流体动力学体积、较小的旋转半径、低结晶度以及低熔融粘度等特点,而这些性质与臂的数目、化学组成、分子量和分子量分布等分子参数密切相关。星形聚合物在许多领域具有潜在的应用价值,如催化、光学、生物工程、涂料、添加剂等,引起了科研工作者们的广泛关注。
然而,合成结构和组成明确且分子量分布较窄的星形聚合物仍然是高分子合成领域一项具有挑战性的工作,目前可用于合成星形聚合物的方法主要有“先臂后核法”和“先核后臂法”。这两种合成路线的基本思路是通过使用多官能团的引发剂或偶联剂、以及双官能团的单体来实现星形聚合物的合成。“先臂后核法”的最大特点是可以方便地设计并控制线型聚合物的分子量,从而得到臂长较为一致的星形聚合物,但其缺点是反应通常需要较长时间,而且很难进行完全;“先核后臂法”的特点是星形聚合物的臂数是由功能性引发基团的数目来确定,而且星形聚合物还可以继续引发单体聚合,获得星形—嵌段共聚物。但这种方法的缺点是,要合成具有明确结构和高纯度的多官能团引发剂相对比较困难;此外,也难以保证每一个引发点的引发效率都保持一致,所以该方法对聚合物臂的分子量和分子量分布等参数的控制性较差。因此,能够快速高效制备结构规整的星形聚合物,并且可以精确控制其组成结构、分子量和分子量分布,仍然是当今高分子合成领域的一个重大挑战;所以也需要研发新的方法用于合成杂臂星形聚合物。
技术实现要素:
本发明的目的在于提供一种八臂杂臂星形聚合物及其制备方法。利用活性阴离子聚合法制备结构精确、分子量分布窄的聚苯乙烯锂化合物,利用其与单羟基七乙烯基多面体齐聚倍半硅氧烷发生加成反应,可以快速制得含单羟基的七臂星形聚合物,并进一步功能化修饰成含叠氮基团的七臂星形聚苯乙烯;再利用它与几种炔基封端的聚合物发生端炔基与叠氮的环加成反应(CuAAC),获得八臂杂臂星形聚合物;本发明公开的方法具有反应效率高、反应条件温和、副反应少、对聚合物的分子量和分子量分布控制好的特点。
为达到上述目的,本发明采取的技术方案为:一种八臂杂臂星形聚合物,其化学结构式如下,
;
式中,R1为(PS);R2选自(PEG)或(PCL)或(PDMA)或(PtBMA);m = 10~40,n = 10~60;*表示连接位点。
优选的,本发明公开的八臂杂臂星形聚合物化学结构式中,R2选自或者。
上述八臂杂臂星形聚合物的制备方法,包括以下步骤:
(1) 制备含单羟基的七臂星形聚苯乙烯:以苯乙烯为单体,仲丁基锂为引发剂,以无水苯或环己烷为溶剂,在20~30℃条件下进行阴离子聚合反应,得到聚苯乙烯锂化合物;然后利用聚苯乙烯锂化合物与单羟基七乙烯基多面体齐聚倍半硅氧烷发生加成反应,得到含单羟基的七臂星形聚苯乙烯;
(2) 制备含叠氮基团的七臂星形聚苯乙烯:惰性气氛中,在三乙胺的催化作用下,以无水四氢呋喃或二氯甲烷为溶剂,利用含单羟基的七臂星形聚苯乙烯和2-溴异丁酰溴在20~50℃发生酯化反应,得到含溴的七臂星形聚苯乙烯;然后含溴的七臂星形聚苯乙烯与叠氮化钠在N, N-二甲基甲酰胺中,于40~80℃发生叠氮反应,得到含叠氮基团的七臂星形聚苯乙烯;
(3) 制备炔基封端的聚己内酯:惰性气氛中,在辛酸亚锡的催化作用下,以无水甲苯为溶剂,在60~80℃条件下,以丙炔醇为引发剂,引发e-己内酯开环聚合反应,得到炔基封端的聚己内酯;
(4) 制备炔基封端的聚乙二醇:惰性气氛中,以聚乙二醇单甲醚为原料,以无水四氢呋喃为溶剂,以氢化钾为催化剂,在20~30℃条件下反应0.5~2小时,然后滴加溴丙炔的四氢呋喃溶液,接着在30~50℃条件下反应12~24小时,得到炔基封端的聚乙二醇;
(5) 制备炔基封端的聚甲基丙烯酸-N, N-二甲氨基乙酯:惰性气氛中,在三乙胺的催化作用下,以无水四氢呋喃或二氯甲烷为溶剂,以丙炔醇和2-溴异丁酰溴为原料,在60~80℃条件下发生酯化反应,制备末端修饰炔基的溴化合物;以末端修饰炔基的溴化合物为引发剂,以溴化亚铜和N, N, N′, N′′, N′′-五甲基二乙烯三胺为催化剂,以无水四氢呋喃或异丙醇为溶剂,在20~40℃条件下引发甲基丙烯酸-N, N-二甲氨基乙酯进行原子转移自由基聚合反应,得到炔基封端的聚甲基丙烯酸-N, N-二甲基氨基乙酯;
(6) 制备炔基封端的聚甲基丙烯酸叔丁酯:惰性气氛中,以步骤(5)制备的末端修饰炔基的溴化合物为引发剂,以溴化亚铜和N, N, N′, N′′, N′′-五甲基二乙烯三胺为催化剂,以无水四氢呋喃或异丙醇为溶剂,在25~40℃条件下引发甲基丙烯酸叔丁酯进行原子转移自由基聚合反应,得到炔基封端的聚甲基丙烯酸叔丁酯;
(7) 制备八臂杂臂星形聚合物:惰性气氛中,以步骤(2)制备的含叠氮基团的七臂星形聚苯乙烯(7PS-POSS-N3)为第一原料,以步骤(3)制备的炔基封端的聚己内酯、步骤(4)制备的炔基封端的聚乙二醇、步骤(5)制备的炔基封端的聚甲基丙烯酸-N, N-二甲氨基乙酯、步骤(6)制备的炔基封端的聚甲基丙烯酸叔丁酯中的一种为第二原料,在端炔基与叠氮的环加成点击反应催化剂和催化剂配体的存在下,以无水四氢呋喃为溶剂,于40~60℃反应12~24小时;得到所述八臂杂臂星形聚合物;
本发明利用阴离子聚合方法可以实现更加快速地制备多臂星形聚合物,并对星形聚合物的结构控制更好;利用羟基与高活性的酰溴发生酯化反应,可以保证羟基完全转化为溴基团,并进一步通过亲核取代反应,转变为叠氮基团;利用点击化学具有反应条件温和、简单高效等特点,能够确保八臂杂臂星形聚合物的结构完整;从而具有反应效率高、反应条件温和、副反应少、对聚合物的分子量和分子量分布控制好的技术效果。
上述技术方案中,具体的制备为:
(1) 制备含单羟基的七臂星形聚苯乙烯(7PS-POSS-OH):以苯乙烯为单体,仲丁基锂为引发剂,以无水苯为溶剂,在20~30℃条件下进行阴离子聚合反应6~12小时,得到聚苯乙烯锂化合物(PSLi);随后利用PSLi与单羟基七乙烯基多面体齐聚倍半硅氧烷(VPOSS-OH)直接发生加成反应0.5~2小时,得到含单羟基的七臂星形聚苯乙烯7PS-POSS-OH;
上述反应式如下:
(2) 制备含叠氮基团的七臂星形聚苯乙烯(7PS-POSS-N3):
(i) 惰性气氛中,在三乙胺的催化作用下,以无水四氢呋喃为溶剂,利用7PS-POSS-OH和2-溴异丁酰溴在20~50℃发生酯化反应12~24小时,得到含溴的七臂星形聚苯乙烯(7PS-POSS-Br);
(ii) 以上述7PS-POSS-Br与叠氮化钠在N, N-二甲基甲酰胺中,于40~80℃反应12~24小时,得到含叠氮基团的七臂星形聚苯乙烯(7PS-POSS-N3);
(3) 制备炔基封端的聚己内酯(Propargyl-PCL):惰性气氛中,在辛酸亚锡的催化作用下,以无水甲苯为溶剂,在60~80℃条件下,以丙炔醇为引发剂,引发e-己内酯开环聚合,反应5~8小时,得到所述的炔基封端的聚己内酯(Propargyl-PCL);
上述反应式如下:
(4) 制备炔基封端的聚乙二醇(Propargyl-PEG):惰性气氛中,以聚乙二醇单甲醚为原料,以无水四氢呋喃为溶剂,以氢化钾为催化剂,在20~30℃条件下反应0.5~2小时,再将溴丙炔溶解在四氢呋喃中,由恒压滴液漏斗中逐滴滴下,在30~50℃条件下反应12~24小时,得到所述的炔基封端的聚乙二醇(Propargyl-PEG);
(5) 制备炔基封端的聚甲基丙烯酸-N, N-二甲氨基乙酯(Propargyl-PDMA)
(i) 惰性气氛中,在三乙胺的催化作用下,以无水四氢呋喃或二氯甲烷为溶剂,以丙炔醇和2-溴异丁酰溴为原料,在60~80℃条件下发生酯化反应,反应12~24小时,制备末端修饰炔基的溴化合物;
上述反应式如下:
(ii) 以上述末端修饰炔基的溴化合物为引发剂,以溴化亚铜和N, N, N′, N′′, N′′-五甲基二乙烯三胺为催化剂,无水四氢呋喃或异丙醇为溶剂,引发甲基丙烯酸-N, N-二甲氨基乙酯进行原子转移自由基聚合,在20~40℃条件下反应6~12小时,得到炔基封端的聚甲基丙烯酸-N, N-二甲基氨基乙酯(Propargyl-PDMA);
(6) 制备炔基封端的聚甲基丙烯酸叔丁酯(Propargyl-PtBMA):惰性气氛中,以末端修饰炔基的溴化合物为引发剂,溴化亚铜和N, N, N′, N′′, N′′-五甲基二乙烯三胺为催化剂,无水四氢呋喃或异丙醇为溶剂,引发甲基丙烯酸叔丁酯进行原子转移自由基聚合,在25~40℃条件下反应6~12小时,得到炔基封端的聚甲基丙烯酸叔丁酯(Propargyl-PtBMA);
(7) 制备八臂杂臂星形聚合物:惰性气氛中,以步骤(2)制备的含叠氮基团的七臂星形聚苯乙烯(7PS-POSS-N3)为第一原料,以步骤(3)制备的炔基封端的聚己内酯、步骤(4)制备的炔基封端的聚乙二醇、步骤(5)制备的炔基封端的聚甲基丙烯酸-N, N-二甲氨基乙酯、步骤(6)制备的炔基封端的聚甲基丙烯酸叔丁酯中的一种为第二原料,在端炔基与叠氮的环加成点击反应催化剂和催化剂配体的存在下,以无水四氢呋喃为溶剂,于40~60℃反应12~24小时;得到所述八臂杂臂星形聚合物。
上述技术方案中,步骤(1)~(7)中各反应物的比例如下:
所述仲丁基锂与苯乙烯的摩尔比为1∶(10~50);
所述PSLi与VPOSS-OH的摩尔比为(8.3~9)∶1;
所述7PS-POSS-OH、2-溴异丁酰溴及三乙胺的摩尔比为1∶(5~20)∶(5~20);
所述7PS-POSS-Br与叠氮化钠的摩尔比为1∶(5~20);
所述丙炔醇、e-己内酯及辛酸亚锡的摩尔比为1∶(50~100)∶(0.25~0.5);
所述聚乙二醇单甲醚、氢化钾及溴丙炔的摩尔比为1∶(2~5)∶(3~5);
所述丙炔醇、2-溴异丁酰溴及三乙胺的摩尔比为1∶(1~1.5)∶(1~1.5);
所述末端修饰炔基的溴化合物、甲基丙烯酸-N, N-二甲氨基乙酯、溴化亚铜及N, N, N′, N′′, N′′-五甲基二乙烯三胺的摩尔比为1∶(10~100)∶1∶1;
所述末端修饰炔基的溴化合物、甲基丙烯酸叔丁酯、溴化亚铜及N, N, N′, N′′, N′′-五甲基二乙烯三胺的摩尔比为1∶(10~100)∶1∶1;
所述第一原料、第二原料、端炔基与叠氮的环加成点击反应催化剂及催化剂配体的摩尔比为1∶(1~1.5)∶(1~2)∶(1~2)。
上述技术方案中,所述步骤(7)中催化剂选自氯化亚铜、溴化亚铜或碘化亚铜;催化剂配体选自联吡啶、五甲基二亚乙基三胺、四甲基乙二胺、六甲基三亚乙基四胺中的一种。
上述技术方案中,所述惰性气氛为氮气或氩气气氛。
上述技术方案中,所述步骤(1)~(7)中,反应完成后,分别对产物进行提纯处理,所述纯化过程包括以下步骤:
1)含单羟基的七臂星形聚苯乙烯(7PS-POSS-OH)的提纯处理:反应结束后,将反应液用旋转蒸发仪浓缩,在甲醇或乙醚中沉淀两次,所得到的产物用环己烷或甲苯溶解至溶液透明状,然后逐渐滴加无水乙醇或甲醇至溶液出现混浊,再将其置于30~35℃水浴中至溶液透明,转移到分液漏斗中,静置,出现分层,取下层透明相,多次反复操作。下层透明相用旋转蒸发仪除去溶剂,浓缩物在甲醇或乙醚中沉淀、抽滤、烘干等操作,得到白色固体粉末,为含单羟基的七臂星形聚苯乙烯;
2)含溴的七臂星形聚苯乙烯(7PS-POSS-Br)的提纯处理:反应结束后,反应液用旋转蒸发仪除去溶剂,再将浓缩产物溶于二氯甲烷溶液中,之后加入饱和碳酸氢钠溶液,振荡后静置,分出下层有机相,水相再用二氯甲烷萃取,合并有机相,有机相溶液经干燥、过滤、浓缩,甲醇或乙醚沉淀得到固体粉末,将所得固体粉末在真空烘箱中20~30℃干燥12~24小时,得到淡黄色固体粉末,为含溴的七臂星形聚苯乙烯;
含叠氮基团的七臂星形聚苯乙烯(7PS-POSS-N3)的提纯处理:末端含溴的星形聚苯乙烯7PS-POSS-Br与叠氮化钠反应结束后,将反应产物过滤、浓缩,再将浓缩液溶于二氯甲烷溶液中,加入饱和氯化钠溶液,振荡后静置,分出下层有机相,水相再用二氯甲烷萃取,合并有机相,有机相溶液经干燥、过滤、浓缩、甲醇或乙醚沉淀得到淡黄色固体粉末,将所得产物在真空烘箱中20~30℃干燥12~24小时,得到含叠氮基团的七臂星形聚苯乙烯;
3) 炔基封端的聚己内酯(Propargyl-PCL)的提纯处理:反应结束后,将反应产物用旋转蒸发仪浓缩,再将浓缩液滴入甲醇或乙醚中沉淀两次、抽滤,将所得产物在真空烘箱中20~30℃干燥12~24小时,得到白色固体,为炔基封端的聚己内酯;
4) 炔基封端的聚乙二醇(Propargyl-PEG)的提纯处理:反应结束后,将反应产物用旋转蒸发仪浓缩,再加入饱和碳酸氢钠溶液,振荡后静置,分出下层有机相,水相再用二氯甲烷萃取,合并有机相,有机相溶液经干燥、过滤、浓缩,在正己烷或乙醚中沉淀两次、抽滤,将所得产物在真空烘箱中20~30℃干燥12~24小时,得到淡黄色固体粉末,为炔基封端的聚乙二醇;
5) 炔基封端的聚甲基丙烯酸-N, N-二甲氨基乙酯(Propargyl-PDMA)的提纯处理:丙炔醇和2-溴异丁酰溴的反应结束后,先过滤除去大部分不溶物,在过滤得到的溶液中加入饱和碳酸氢钠溶液,振荡后静置,分出下层有机相,水相再用二氯甲烷萃取,合并有机相,有机相溶液经干燥、过滤、浓缩,并在真空烘箱中20~30℃干燥12~24小时,得到无色透明状末端修饰炔基的溴化合物;该小分子与甲基丙烯酸-N, N-二甲氨基乙酯反应结束后,将反应混合物流经硅胶柱,用旋转蒸发仪浓缩,在正己烷或乙醚中沉淀两次、抽滤,将所得产物在真空烘箱中20~30℃干燥12~24小时,得到淡黄色透明黏状固体。
6) 炔基封端的聚甲基丙烯酸叔丁酯(Propargyl-PtBMA)的提纯处理:与甲基丙烯酸叔丁酯反应结束后,将反应混合物流经硅胶柱,用旋转蒸发仪浓缩,在正己烷或乙醚中沉淀两次、抽滤,将所得产物在真空烘箱中20~30℃干燥12~24小时,得到白色透明黏状固体。
7) 八臂杂臂星形聚合物的提纯处理:反应结束后,将反应液过硅胶柱,用甲苯冲洗,再经过旋转蒸发仪浓缩、甲醇沉淀,得到淡黄色固体粉末,并将淡黄色固体粉末在真空烘箱中20~30℃干燥12~24小时,为八臂杂臂星形聚合物。
优选的,步骤(2)中,惰性气体氛围下,在-5~0℃下,将含单羟基的七臂星形聚苯乙烯的四氢呋喃溶液滴加入2-溴异丁酰溴的四氢呋喃溶液中,然后在20~50℃发生酯化反应,得到含溴的七臂星形聚苯乙烯。
本发明公开的方法具有反应效率高、反应条件温和、副反应少、对聚合物的分子量和分子量分布控制好的优点,得到的八臂杂臂星形聚合物结构可控、性能良好,可用于多种用途;由于含有PEG和PDMA链段的八臂星形聚合物具有很好的两亲性质,可以在水溶液中组装形成胶束,其内核用于负载疏水性分子(药物、染料等),在载体材料和体外诊断试剂有潜在的应用;此外,由于八臂星形聚合物具有较高的热分解温度,可以在涂料添加剂中有潜在的应用。因此本发明还公开了上述八臂杂臂星形聚合物在制备载体材料、体外诊断试剂、耐热涂料中的应用。
由于上述方案运用,本发明与现有技术相比,具有以下优点:
1.本发明首次采用一种高效模块化组合思路,将活性阴离子聚合和端炔基与叠氮的环加成反应(CuAAC)联用,合成了多种八臂杂臂星形聚合物。
2. 本发明利用活性阴离子聚合法制得的聚苯乙烯活性链直接与单羟基七乙烯基多面体齐聚倍半硅氧烷发生加成反应,可以快速高效地制备含羟基的七臂星形聚苯乙烯前体,采用活性阴离子聚合法可以方便高效地控制聚合物链的分子量和分子量分布。
3. 本发明将七臂星形聚苯乙烯前体中的羟基转变为叠氮基团,并利用它与各种末端含炔基的聚合物发生端炔基与叠氮的环加成反应(CuAAC),可以高效地合成化学结构多样化的杂臂星形聚合物。
附图说明
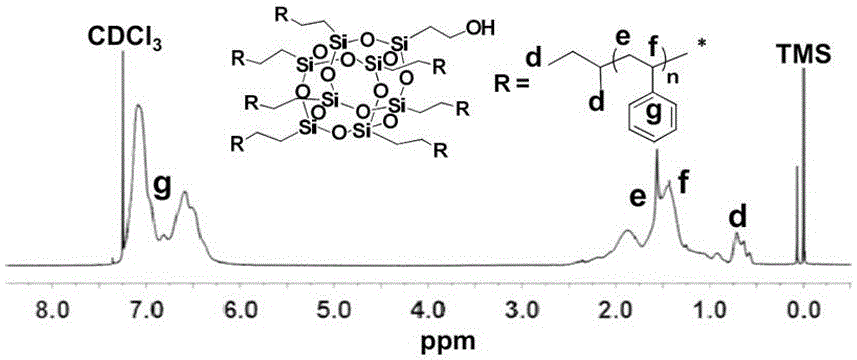
图1为实施例一中7PS-POSS-OH的核磁共振氢谱图,溶剂为氘代氯仿(CDCl3);
图2为实施例一中PS和7PS-POSS-OH的凝胶渗透色谱流出曲线,溶剂为四氢呋喃(THF);
图3为实施例一中7PS-POSS-OH的红外光谱谱图;
图4为实施例一中Propargyl-PCL的大分子质谱图;
图5为实施例一中7PS-POSS-N3(A)、Propargyl-PCL(B)和7PS-POSS-PCL(C)的核磁共振氢谱图,溶剂为氘代氯仿(CDCl3);
图6为实施例一中7PS-POSS-N3(A)、Propargyl-PCL(B)和7PS-POSS-PCL(C)的红外光谱图;
图7为实施例一中7PS-POSS-N3、Propargyl-PCL和7PS-POSS-PCL的凝胶渗透色谱流出曲线,溶剂为四氢呋喃(THF);
图8为实施例一中7PS-POSS-PCL的热失重曲线;
图9为实施例二中Propargyl-PEG的大分子质谱图;
图10为实施例二中7PS-POSS-N3(A)、Propargyl-PEG(B)、7PS-POSS-PEG(C)的核磁共振氢谱图,溶剂为氘代氯仿(CDCl3);
图11为实施例二中7PS-POSS-N3(A)、Propargyl-PEG(B)和7PS-POSS-PEG(C)的红外光谱图;
图12为实施例二中7PS-POSS-N3、Propargyl-PEG和7PS-POSS-PEG的凝胶渗透色谱流出曲线,溶剂为四氢呋喃(THF);
图13为实施例二中7PS-POSS-PEG的热失重曲线。
具体实施方式
下面结合附图及实施例对本发明作进一步描述:
实施例一:八臂杂臂星形聚合物(7PS-POSS-PCL)的制备
(1)含单羟基的七臂星形聚苯乙烯(7PS-POSS-OH)的制备
用仲丁基锂(sec-BuLi,3.8 mL,4.96 mmol)作为引发剂,苯(50 mL)作溶剂,在30℃条件下引发苯乙烯(7.8 mL, 68.1 mmol)进行活性阴离子聚合,反应8小时后取少量活性链溶液用甲醇终止,剩余的活性链在真空条件下继续与VPOSS-OH(389.3 mg, 0.6 mmol)直接发生加成反应,反应1小时后用甲醇终止。
反应结束后,将产物进行旋蒸除去溶剂,并在甲醇中进行沉淀,过滤、浓缩,得到白色固体粉末,将所得固体粉末在真空烘箱中常温干燥24小时,得到7PS-POSS-OH粗产物,再用乙醇/环己烷作为溶剂,通过分级沉淀得到纯的7PS-POSS-OH,产率为32%。
利用核磁共振氢谱(1H NMR)、凝胶渗透色谱(GPC)和红外光谱(FT-IR)对其进行表征。附图1、附图2和附图3分别为上述7PS-POSS-OH的核磁共振氢谱(1H NMR)图、凝胶渗透色谱(GPC)流出曲线和红外光谱(FT-IR)图,验证了7PS-POSS-OH的化学结构。从1H NMR谱图中可以找到对应于聚合物结构上的质子峰归属,从GPC流出曲线(PS, = 1460 g•mol-1, = 1.09;7PS-POSS-OH, = 14350 g•mol-1, = 1.11)中可以看到纯化得到的聚合物的峰形比较对称,分子量分布比较窄。
(2)含叠氮基团的七臂星形聚苯乙烯(7PS-POSS-N3)的制备
惰性气体氛围下,在支管烧瓶里加入一定量的2-溴异丁酰溴(0.62 g, 2.68 mmol),并用少量的THF溶解搅拌。将7PS-POSS-OH(1.48 g, 0.134 mmol)用THF溶解并加入到恒压滴液漏斗中,然后用注射器加入一定量的TEA。在冰水浴条件(-5~0℃)下,使恒压滴液漏斗中的溶液逐滴加入到支管瓶中,滴加结束后将整个装置置于30℃的油浴中反应24 h。反应完毕,旋蒸除去溶剂,继续用二氯甲烷溶解,并用饱和NaHCO3萃取两遍,每次约30 mL,再用约30 mL饱和NaCl萃取至溶液澄清透明,最后用无水Na2SO4干燥,过滤,旋干,用甲醇沉淀,在真空烘箱中干燥得到淡黄色粉末。
继续将所得到的固体溶于10 mL的DMF中,并称取NaN3(0.13 g, 2.69 mmol)于溶液中,在30℃条件下反应24 h,待反应结束后,除去DMF,沉淀以及干燥即可得到淡黄色粉末状固体7PS-POSS-N3,产率65.5%。
利用核磁共振氢谱(1H NMR)、红外光谱(FT-IR)和凝胶渗透色谱(GPC)对其进行表征。附图5(A)、附图6(A)和附图7分别为上述7PS-POSS-N3的核磁共振氢谱(1H NMR)图、红外光谱(FT-IR)图及凝胶渗透色谱(GPC)流出曲线,验证了7PS-POSS-N3的化学结构。从1H NMR谱图中可以找到对应于聚合物结构上的质子峰归属,从GPC流出曲线(7PS-POSS-N3, = 15130 g•mol-1, = 1.12)中可以看到纯化得到的聚合物的峰形比较对称,分子量分布比较窄,从FT-IR谱图中可以看到明显的叠氮基团的特征峰(2105 cm-1)。
(3)炔基封端的聚己内酯(Propargyl-PCL)的制备
惰性气氛中,以无水甲苯为溶剂,依次将丙炔醇(100 mg, 1.78 mmol),e-己内酯(10.17 g, 89.12 mmol)以及辛酸亚锡(0.9 mL, 0.45 mmol, 0.5 M solution in Toluene)通过干燥好的注射器加入到反应瓶中进行开环聚合,反应6小时,过滤、浓缩并用甲醇沉淀,在真空烘箱中干燥得到所述的炔基封端的聚己内酯,产率19.7%。
利用核磁共振氢谱(1H NMR)、红外光谱(FT-IR)、凝胶渗透色谱(GPC)、大分子质谱(MALDI-TOF MS)和对其进行了表征。附图4、附图5(B)、附图6(B)和附图7分别为上述Propargyl-PCL的大分子质谱图(MALDI-TOF MS)、核磁共振氢谱(1H NMR)图、红外光谱(FT-IR)图和凝胶渗透色谱(GPC)流出曲线,验证其化学结构。从1H NMR谱图中可以找到对应于聚合物结构上的质子峰归属,从GPC流出曲线(Propargyl-PCL,= 3360 g•mol-1,= 1.08)中可以看到纯化得到的聚合物的峰形比较对称,分子量分布比较窄,从FT-IR谱图中可以看到明显的炔基基团(3280 cm-1)和羰基基团(1730 cm-1)的特征峰。
(4)八臂杂臂星形聚合物7PS-POSS-PCL的制备
惰性气体氛围下,向圆底瓶中加入7PS-POSS-N3(100 mg, 0.009 mmol)和炔基封端的聚己内酯(20.15 mg, 0.011 mmol),并用10 mL的THF溶解。快速加入CuBr(2.58 mg, 0.018 mmol)和PMDETA(3.12 mg, 0.018 mmol),在45℃油浴下反应24 h。反应结束后,将溶液流经硅胶柱,再经过旋蒸、沉淀、干燥,得到淡黄色固体粉末7PS-POSS-PCL, 产率70%。
附图5(C)、附图6(C)和附图7分别为上述7PS-POSS-PCL的核磁共振氢谱(1H NMR)图、红外光谱(FT-IR)图和凝胶渗透色谱(GPC)流出曲线,验证了7PS-POSS-PCL的化学结构以及八臂杂臂星形聚合物的成功合成。从1H NMR谱图中可以找到对应于星形聚合物结构上的质子峰归属,从GPC流出曲线(7PS-POSS-PCL, = 16500 g•mol-1, = 1.13)中可以看到纯化得到的星形聚合物的峰形比较对称,分子量分布比较窄。附图8为上述7PS-POSS-PCL及其聚合物前体的热失重(TGA)曲线,从图中可以看出星形聚合物具有较好的热稳定性。
实施例二:八臂杂臂星形聚合物(7PS-POSS-PEG)的制备
(1)炔基封端的聚乙二醇(Propargyl-PEG)的制备
向100 mL支管烧瓶加入一定量的mPEG-OH(2.00 g, 1 mmol)和40 mL无水甲苯,共沸除水。结束后在惰性气体氛围下将纯化的mPEG-OH用无水THF溶解,缓慢加入KH(0.24 g, 6 mmol),并在25℃油浴条件下搅拌反应1 h。
用注射器向恒压滴液漏斗中加入3-溴丙炔(1.19 g, 10 mmol, 80% in Toluene),并用约5 mL的THF稀释。紧接着逐滴加入到支管瓶中,待滴加完毕后在45℃油浴下反应20 h。过滤、浓缩后加入40 mL的CH2Cl2,用饱和NaHCO3溶液萃取三次,收集有机相,用无水Na2SO4干燥12 h,过滤旋干,用正己烷沉淀,在真空烘箱中干燥后得到淡黄色的炔基封端PEG,产率91%。
利用核磁共振氢谱(1H NMR)、凝胶渗透色谱(GPC)、大分子质谱(MALDI-TOF MS)和红外光谱(FT-IR)对其进行表征。附图9、附图10(B)、附图11(B)和附图12分别为上述Propargyl-PEG的大分子质谱图(MALDI-TOF MS)、核磁共振氢谱(1H NMR)图、红外光谱(FT-IR)图和凝胶渗透色谱(GPC)流出曲线,验证了其化学结构。从1H NMR谱图中可以找到对应于聚合物结构上的质子峰归属,从GPC流出曲线(Propargyl-PEG, = 3000 g•mol-1, = 1.05)中可以看到纯化得到的聚合物的峰形比较对称,分子量分布比较窄,从FT-IR谱图中可以看到明显的炔基基团(3280 cm-1)的特征峰。
(2)八臂杂臂星形聚合物7PS-POSS-PEG的制备
惰性气体氛围下,向圆底瓶中加入实施例一制得的7PS-POSS-N3(100 mg, 0.009 mmol)和Propargyl-PEG(26.5 mg, 0.0126mol),并用12 mL的THF溶解。快速入CuBr(2.0 mg, 0.0135mmol)和PMDETA(2.34 mg, 0.0135 mmol),在55℃油浴下反应22 h。反应结束后,将溶液流经硅胶柱,再经过旋蒸、沉淀、干燥,得到淡黄色固体粉末7PS-POSS-PEG, 产率66%。
附图10(C)、附图11(C)和附图12分别为上述7PS-POSS-PEG的核磁共振氢谱(1H NMR)图、红外光谱(FT-IR)图和凝胶渗透色谱流出曲线(GPC,以7PS-POSS-N3为参考),验证了7PS-POSS-PEG的化学结构以及八臂杂臂星形聚合物的成功合成;其中图10(A)、图11(A)以7PS-POSS-N3为参考。从1H NMR谱图中可以找到对应于星形聚合物结构上的质子峰归属,从GPC流出曲线(7PS-POSS-PEG, = 16170 g•mol-1, = 1.11)中可以看到纯化得到的星形聚合物的峰形比较对称,分子量分布比较窄。附图13为上述7PS-POSS-PEG及其聚合物前体的热失重(TGA)曲线,从图中可以看出星形聚合物具有较好的热稳定性。
实施例三:八臂杂臂星形聚合物(7PS-POSS-PDMA)的制备
(1)含单羟基的七臂星形聚苯乙烯(7PS-POSS-OH)的制备
用仲丁基锂(sec-BuLi,3.0 mL,3.92 mmol)作引发剂,苯(40 mL)作为溶剂,在30℃条件下引发苯乙烯(8.16 mL, 78.4 mmol)进行活性阴离子聚合,反应10小时后取少量活性链溶液用甲醇终止,剩余的活性链在真空条件下继续与VPOSS-OH(259.5 mg, 0.4 mmol)直接发生加成反应,反应1小时后用甲醇终止。
反应结束后,将产物进行旋蒸除去溶剂,并在甲醇中进行沉淀,过滤、浓缩,得到白色固体粉末,将所得固体粉末在真空烘箱中常温干燥24小时,得到7PS-POSS-OH粗产物,再用乙醇/环己烷作为溶剂,通过分级沉淀得到纯的7PS-POSS-OH,产率为35%。
(2)含叠氮基团的七臂星形聚苯乙烯(7PS-POSS-N3)的制备
惰性气体氛围下,在支管烧瓶里加入一定量的2-溴异丁酰溴(0.35 g, 1.5 mmol),并用少量的THF溶解搅拌。将7PS-POSS-OH(1.60 g, 0.1 mmol)用THF溶解并加入到恒压滴液漏斗中,然后用注射器加入一定量的TEA。在冰水浴条件(-5~0℃)下,使恒压滴液漏斗中的溶液逐滴加入到支管瓶中,滴加结束后将整个装置置于35℃的油浴中反应20 h。反应完毕,旋蒸除去溶剂,继续用二氯甲烷溶解,并用饱和NaHCO3萃取两遍,每次约35 mL,再用约35 mL饱和NaCl萃取至溶液澄清透明,最后用无水Na2SO4干燥,过滤,旋干,用甲醇沉淀,在真空烘箱中干燥得到淡黄色粉末。
继续将所得到的固体溶于10 mL的DMF中,并称取NaN3(0.097 g,2.0 mmol)于溶液中,在30℃条件下反应24 h,待反应结束后,除去DMF,沉淀以及干燥即可得到淡黄色粉末状固体7PS-POSS-N3,产率70%。
(3)炔基封端的聚甲基丙烯酸-N, N-二甲氨基乙酯(Propargyl-PDMA)的制备
惰性气体氛围下,在支管瓶中加入CuBr(21.5 mg,0.15 mmol),抽真空, 然后用针筒依次注入末端修饰炔基的溴化合物(30.8 mg,0.15 mmol),DMA单体(0.943 g,6.0 mmol),PMDETA(26 mg,0.15 mmol)以及10 mL THF,在35℃搅拌反应6小时。反应结束后,将溶液流经硅胶柱,再经过旋蒸、正己烷沉淀、30℃真空烘箱干燥12小时,即可得到淡黄色黏状固体Propargyl-PDMA,产率65%。
(4)八臂杂臂星形聚合物7PS-POSS-PDMA的制备
惰性气体氛围下,向圆底瓶中加入7PS-POSS-N3(160 mg, 0.01 mmol)和Propargyl-PDMA(52.5 mg, 0.015 mmol),并用15 mL的THF溶解。快速入CuCl(2.985 mg, 0.03 mmol)和PMDETA(5.2 mg, 0.03 mmol),在50℃油浴下反应24 h。反应结束后,将溶液流经硅胶柱,再经过旋蒸、沉淀、干燥,得到淡黄色固体粉末7PS-POSS-PDMA,产率64%。
实施例四:八臂杂臂星形聚合物(7PS-POSS-PtBMA)的制备
(1)炔基封端的聚甲基丙烯酸叔丁酯(Propargyl-PtBMA)的制备
惰性气体氛围下,在支管瓶中加入CuBr(21.5 mg,0.15 mmol),抽真空, 然后用针筒依次注入末端修饰炔基的溴化合物(30.8 mg,0.15 mmol),tBMA单体(0.853 g,6.0 mmol),PMDETA(26 mg,0.15 mmol)以及10 mL THF,在35℃搅拌反应6小时。反应结束后,将溶液流经硅胶柱,再经过旋蒸、正己烷沉淀、30℃真空烘箱干燥12小时,即可得到淡黄色黏状固体Propargyl-PDMA,产率70%。
(2)八臂杂臂星形聚合物7PS-POSS-PtBMA的制备
惰性气体氛围下,向圆底瓶中加入实施例一制得的7PS-POSS-N3(100 mg, 0.009 mmol)和Propargyl-PtBMA(54.6 mg, 0.013 mmol),并用10 mL的THF溶解。快速入CuBr(1.94 mg, 0.0135 mmol)和PMDETA(2.34 mg, 0.0135 mmol),在50℃油浴下反应20 h。反应结束后,将溶液流经硅胶柱,再经过旋蒸、沉淀、干燥,得到淡黄色固体粉末7PS-POSS-PtBMA,产率60%。
- 还没有人留言评论。精彩留言会获得点赞!