丝状真菌中多个基因拷贝的同时位点特异性整合的制作方法
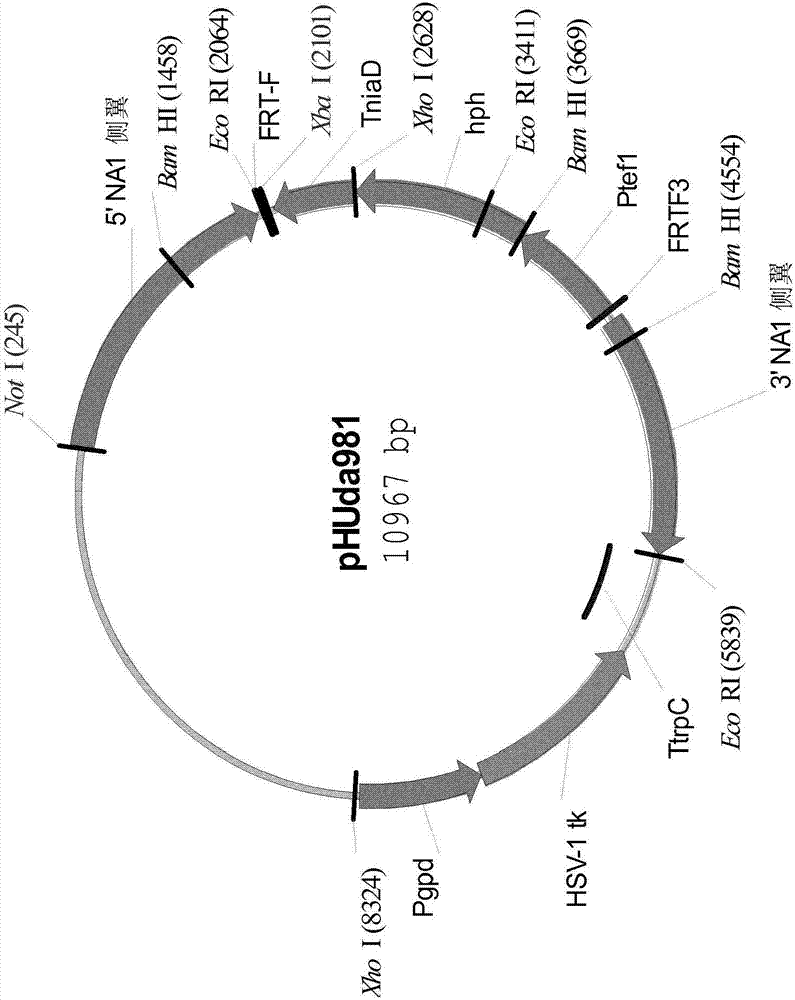
本申请是2013年11月25日提交的发明名称为“丝状真菌中多个基因拷贝的同时位点特异性整合”、申请号为“201280025301.9”的发明专利申请的分案申请。对序列表的引用本申请包含计算机可读形式的序列表。该计算机可读形式并入本文以作参考。本发明涉及使用短暂表达的重组酶连同合适的驻留(resident)选择标记将多个拷贝的目标多核苷酸同时位点特异性地整合到真菌宿主细胞的基因组中的方法。
背景技术:
:已经发现大量的自然产生的生物体产生有用的多肽产物,例如酶,酶的大规模生产为研究和商业用途所需。在鉴定出这样的多肽产物后,经常要致力于开发具有改进的生产率的制造方法。一种广泛运用的基于重组dna技术的方法是克隆编码该产物的基因,并将该基因插入到合适的表达体系中,从而在有益于产物表达的条件下在合适的宿主细胞中表达该产物,该基因或是整合到染色体中,或是作为染色体外的实体。无论使用何种生产方法,通常期望提高给定的多肽或蛋白质的生产水平。因此,正在为增加生产而作出努力,例如,通过将编码该产物的基因插入到强表达信号的控制下,增加所转录的mrna的稳定性或通过增加所考虑的生产生物体中该基因的拷贝数。后一种方法可以通过将该基因插入到多拷贝质粒中来实现,但是通常多拷贝质粒存在在所考虑的宿主细胞中不稳定的倾向,或者通过将多个拷贝的该基因整合到生产生物体的染色体中来实现,该途径由于构建体的稳定性趋向于是更高的而通常被认为是更具有吸引力的。宿主细胞的构建已被描述过,其中高度表达的染色体基因由位点特异性重组酶的识别序列替代,从而可以在随后通过使用识别所述序列的重组酶将单个编码产物的多核苷酸插入到那个位点(ep1405908a1;probiogenag)。已经公开了在基因组中的已知位置插入dna(o'gorman等,1991science,251:1351-55;baubonis和sauer,1993nucl.,acidsres.,21:2025-29;albert等,1995plantj.,7:649-59)。这些方法利用自由可逆的位点特异性重组体系。这些可逆体系包括以下:来自噬菌体p1的cre-lox体系(baubonis和sauer,1993,见上文;albert等,1995plantj.,7549-59),酿酒酵母(saccharomycescerevisiae)的flp-frt体系(o'gorrnan等,1991,见上文),zygosaccharonzycesrouxii的r-rs体系(onouchi等,1995mol.gen.genet.247:653-660),来自mu噬菌体的改良gin-gix体系(maeser和kahmann,1991mol.gen.genet.,230:170-76),来自枯草芽孢杆菌(bacillussubtilis)质粒的β-重组酶-six体系(diaz等,1999j.biol.chem.274:6634-6640)以及来自细菌转座子tn1000的δ-γ-res体系(schwikardi和dorge,2000ebslet.471:147-150)。cre、flp、r、gin、β-重组酶和δ-γ是重组酶,lox、frt、rs、gix、six和res是各个重组位点(由sadowslu,1993fasebj.,7:750-67;ow和medberry,1995crit.rev.plantsci.14:239-261综述)。多重cre/lox重组允许选择性位点特异性dna靶向酵母基因组中的自然位点和工程化位点(sauer,b.nucleicacidsresearch.1996,vol.24(23):4608-4613)。已经显示使用链霉菌属(streptomyces)噬菌体φc31的衍生物感染具有自然附着位点attb和异位引入的attb位点的宿主细胞,结果使该噬菌体整合到两个attb位点中(smith等,2004,switchingthepolarityofabacteriophageintegrationsystem.molmicrobiol51(6):1719-1728)。使用mx9噬菌体转化体系,多个拷贝的基因可以引入到包含多个被mx9整合酶识别的附着位点的细胞中(wo2004/018635a2)。温带lactococcal噬菌体tp901-1整合酶和识别序列是良好表征的(breüner等,(1990)novelorganizationofgenesinvolvedinprophageexcisionidentifiedinthetemperatelactococcalbacteriophagetp901-1.jbacteriol181(23):7291-7297;breüner等,2001.resolvase-likerecombinationperformedbythetp901-1integrase.microbiology147:2051-2063)。以上的位点特异性重组体系具有共同的性质,即单一的多肽重组酶催化相同序列或近乎相同序列在两个位点间的重组。每个重组位点由短的不对称间隔序列组成,在该短的不对称间隔序列处发生链交换,该短的不对称间隔序列在两侧有重组酶结合的反向重复序列。间隔序列的不对称性对重组位点提供了一个方向,并规定了重组反应的结果。直接或间接定向的顺式位点之间的重组分别切除或倒置干预dna。反式位点之间的重组引起两个线性dna分子的相互易位,或者,如果两个分子之中有至少一个是环形的,则引起共整合。因为由重组产生的产物位点自身是随后重组的底物,所以反应是自由可逆的。但是,在实际中,因为两个重组位点紧密相连的地方的分子内相互作用的可能性远远高于不相连位点间的分子内相互作用的可能性,所以切除基本上是不可逆的。必然的结果是插入到基因组重组位点中的dna分子会很容易地切除掉。已经公开了在真核生物的基因组中进行dna取代、易位和叠加的方法,在这些方法中多个基因可以一步步整合(wo02/08409)。通过位点特异性和短暂表达的整合酶在微生物宿主细胞中进行多个拷贝的无启动子开放阅读框或操纵子的同时基因组整合(wo2006/042548)早前已经在芽孢杆菌(bacillus)宿主中示出。技术实现要素:本发明涉及将两个或更多个拷贝的目标多核苷酸整合到真菌宿主细胞的染色体中的方法,该方法通过以下步骤进行:(a)提供在其染色体内包括至少两个整合位点的真菌宿主细胞,每个整合位点包括一对位点特异性重组酶的识别序列,每对识别序列位于驻留(resident)选择标记的两侧;(b)向所述细胞中引入核酸构建体,该核酸构建体包括一对位点特异性重组酶的识别序列,该识别序列对位于目标多核苷酸的两侧;(c)在细胞中短暂表达位点特异性重组酶,借此染色体的识别序列对通过重组酶与核酸构建体的对应的识别序列对重组,从而使得在至少两个整合位点,染色体中的驻留选择标记被切除而目标多核苷酸的拷贝整合到它的位置上,产生包括两个或更多个拷贝的整合到真菌宿主细胞的染色体中的目标多核苷酸的真菌宿主细胞。如在以下的实施例部分中所例证的,本发明的主要方面提供将两个或更多个拷贝的目标多核苷酸同时整合到真菌宿主细胞的染色体中的方法,该方法包括以下步骤:(a)提供在其染色体内包括至少两对位点特异性重组酶的识别序列的真菌宿主细胞,每对识别序列位于驻留阴性选择标记的两侧;(b)向所述细胞中引入核酸构建体,该核酸构建体包括一对位点特异性重组酶的识别序列,该识别序列对位于目标多核苷酸的两侧;(c)在细胞中短暂表达位点特异性重组酶,借此染色体的识别序列对通过重组酶与核酸构建体的对应的识别序列对重组,从而使得染色体中的每个驻留阴性选择标记被切除而目标多核苷酸的拷贝整合到它的位置上;然后(d)在选择性培养基中培养该细胞并选择细胞,其中每个阴性选择标记已经通过双同源重组被两个或更多个拷贝的目标多核苷酸取代。附图说明图1显示本发明方法的基本方案,在此运用frt/flp重组体系和pyrg(作为双向选择标记)以及两侧是两拷贝转录终止子(标记为“term”)的flp编码基因一起加以例证,其使得可以通过双同源重组实现flp和pyrg基因的切除,使得可以进行flp的短暂表达。图2显示phuda981的质粒图谱(在wo07045248中描述了pgpd,hsv1tk和ttrpc)。图3显示phuda1019的质粒图谱。图4显示phuda1000的质粒图谱。图5显示phuda1000正确引入到nn059183中之后的na1(上图)和酸稳定性淀粉酶基因座示意图(下图)。图6显示phuda801的质粒图谱。图7显示phuda1043的质粒图谱。图8显示phuda1078的质粒图谱。图9显示phuda1067的质粒图谱。图10显示nn059208中的na1基因座(上部)、na2基因座(中间)和酸稳定性淀粉酶基因座(下部)示意图。图11显示prika147的质粒图谱。图12显示prika147正确整合到nn059208中之后的na1(上部)、na2(中间)和酸稳定性淀粉酶基因座(下部)示意图。图13显示phuda1174的质粒图谱。图14显示m1146中pay基因座(上部)的示意图。图15显示phuda1306的质粒图谱。图16a显示当prika147引入到m1146中时的na1基因座(上部)和na2(2nd)的示意图。图16b显示当prika147引入到m1146中时的sp288基因座(3rd)示意图。图16c显示当prika147引入到m1146中时的pay基因座示意图。图17显示phuda1356的质粒图谱。图18显示用来将frt位点在cbh1基因座处整合到里氏木霉(t.reesei)基因组中的载体pjfys147的图谱。图19显示使用flp/frt体系用来将烟曲霉(a.fumigatus)bg在cbh1基因座处整合到里氏木霉菌株jfys147-20b中的载体pjfys150的图谱。定义胞嘧啶脱氨酶:胞嘧啶脱氨酶(ec3.5.4.1)催化胞嘧啶和5-氟胞嘧啶(5fc)的脱氨基作用以分别形成尿嘧啶和有毒的5-氟尿嘧啶(5fu)。当包含胞嘧啶脱氨酶的基因改造细胞与5fc结合时,5fc被转化成有毒的5fu,因此胞嘧啶脱氨酶编码基因潜在地是有效的阴性选择标记。也已显示,嘧啶从头(denovo)合成途径中的抑制剂可以用于创造如下条件,在该条件下细胞依赖于通过胞嘧啶脱氨酶进行的嘧啶补充物到尿嘧啶的转化。因此,只有表达胞嘧啶脱氨酶基因的细胞可以在包含嘧啶从头合成抑制剂及肌苷和胞嘧啶的阳性选择介质中被救援(参见weiandhuber,1996,jbiolchem271(7):3812的图1)。该抑制剂优选是n-膦乙酰基-l-天冬氨酸(pala),其抑制天冬氨酸氨甲酰基转移酶。如果必要的话,胞嘧啶脱氨酶活性可以通过基因检测加以定量(fredericol.a.等,1990,biochemistry29:2532-2537)。等位变体(allelicvariant):术语“等位变体”是指占据相同染色体基因座的两种或多种可替换基因形式中的任一种。等位变体自然地通过突变而产生,且可以引起群体内的多态性。基因突变可以是沉默的(在编码的多肽中没有变化),或者可以编码具有改变的氨基酸序列的多肽。多肽的等位变体是由基因的等位变体编码的多肽。催化结构域:术语“催化结构域”是指酶的包含酶的催化机构的区域。cdna:术语“cdna”是指能够通过得自真核或原核细胞的成熟的、剪接的mrna分子的反转录而制得的dna分子。cdna缺少可能存在于相应基因组dna中的内含子序列。早先的初始rna转录子是mrna的前体,其在呈现为成熟的剪接的mrna之前要经一系列的步骤加工,包括剪接。编码序列:术语“编码序列”是指多核苷酸,其直接明确多肽的氨基酸序列。编码序列的边界通常由开放阅读框决定,其以起始密码子例如atg、gtg或ttg开始,并以终止密码子例如taa、tag或tga结束。编码序列可以是基因组dna、cdna、合成dna或其组合。控制序列:术语“控制序列”是指对于本发明的编码成熟多肽的多核苷酸的表达为必要的核酸序列。各个控制序列可以相对于编码多肽的多核苷酸是原生的(native)(即,来自相同基因)或外源的(即,来自不同基因)或者相互为原生的或外源的。这样的控制序列包括但不限于,前导序列、多腺苷酸化序列、前肽序列、启动子、信号肽序列和转录终止子。控制序列至少包括启动子、以及转录和翻译终止信号。出于引入特定限制性酶切位点的目的,控制序列可具有接头,其促进控制序列与编码多肽的多核苷酸的编码区域的连接。表达:术语“表达”包括涉及到多肽生产中的任何步骤,包括但不限于:转录、转录后修饰、翻译、翻译后修饰以及分泌。表达载体:术语“表达载体”是指包括编码多肽的多核苷酸并且可操作地与用于其表达的控制序列相连接的线性或环状dna分子。片段:术语“片段”是指具有一个或多个(例如,数个)从成熟多肽或结构域的氨基和/或羧基端缺失的氨基酸的多肽或催化结构域;其中片段具有胞嘧啶脱氨酶活性。宿主细胞:术语“宿主细胞”是指对转化、转染、转导等敏感的任何细胞类型,其具有包含本发明的多核苷酸的核酸构建体或表达载体。术语“宿主细胞”包括因复制过程中发生的突变而与亲本细胞不同的亲本细胞的任意后代。分离的或纯化的:术语“分离的”或“纯化的”是指从与其自然相关联的至少一个成分中移出的多肽或多核苷酸。例如,按照sds-page所测定的,多肽可以是至少1%纯的,例如至少5%纯,至少10%纯,至少20%纯,至少40%纯,至少60%纯,至少80%纯,至少90%纯,或至少95%纯,如按照琼脂糖电泳所测定的,多核苷酸可以是至少1%纯的,例如至少5%纯,至少10%纯,至少20%纯,至少40%纯,至少60%纯,至少80%纯,至少90%纯,或至少95%纯。阴性选择标记:术语“阴性选择标记”是指能够赋予选择特征从而使得具有该阴性选择标记的细胞被杀死或者以其他方式例如通过荧光被识别的核酸序列。阴性选择标记优选基本不能与靶标dna序列同源重组。核酸构建体:术语“核酸构建体”是指从天然存在的基因中分离的、或以自然中不会另外出现的方式被修饰成包含核酸片段的、或合成的单链或双链的核酸分子,其包括一个或多个控制序列。可操作地连接:术语“可操作地连接”是指如下的构造,其中,控制序列相对于多核苷酸的编码序列安置在合适位置处,从而控制序列指导编码序列的表达。序列同一性:两个氨基酸序列之间或两个核苷酸序列之间的关联性通过参数“序列同一性”来说明。出于本发明的目的,使用在emboss软件包(emboss:theeuropeanmolecularbiologyopensoftwaresuite(欧洲分子生物学开放软件包),rice等人,2000,trendsgenet.16:276-277)(优选5.0.0版本或更新)的needle程序中执行的needleman-wunsch算法(needlemanandwunsch,1970,j.mol.biol.48:443-453)来确定两个氨基酸序列之间的序列同一性。所用的参数:空位开放罚分为10,空位扩展罚分为0.5,以及eblosum62(blosum62的emboss版本)替换矩阵。needle标记的“最长一致度”的输出(使用-nobrief选项得到的)被用作百分比一致度并如下计算:(相同残基×100)/(比对长度-比对中的空位总数)出于本发明的目的,使用在emboss软件包(emboss:theeuropeanmolecularbiologyopensoftwaresuite(欧洲分子生物学开放软件包),rice等人,2000,同上)(优选5.0.0版本或更新)的needle程序中执行的needleman-wunsch算法(needlemanandwunsch,1970,同上)来确定两个脱氧核糖核苷酸序列之间的序列同一性。所用的参数:空位开放罚分为10,空位扩展罚分为0.5,以及ednafull(ncbinuc4.4的emboss版本)替换矩阵。needle标记的“最长一致度”的输出(使用-nobrief选项得到的)被用作百分比一致度并如下计算:(相同脱氧核糖核酸×100)/(比对长度-比对中的空位总数)同时:本文中所使用的术语“同时”,即,关于至少两个目标多核苷酸向宿主细胞中的整合,指的是这样一个过程,通过这个过程在至少两个拷贝的目标多核苷酸向宿主细胞中的整合发生在同一过程步骤中,致使增加一个拷贝的目标多核苷酸,即,不需要增加任何其它材料和/或任何附加过程步骤。因此,目标多核苷酸在同一过程中在相同时刻或不同时刻但在同一过程中同时期地被引入到该宿主细胞中的至少两个不同整合位点。子序列:术语“子序列”是指从成熟多肽编码序列的5’和/或3’端缺失一个或多个(例如,数个)核苷酸的多核苷酸;其中子序列编码具有胞嘧啶脱氨酶活性的片段。变体:术语“变体”是指在一个或多个位置上包括一个或多个(例如,数个)氨基酸残基的改变(即替换、插入和/或缺失)的具有胞嘧啶脱氨酶活性的多肽。替换是指用不同的氨基酸替换占据某位置的氨基酸;缺失是指去除占据位置的氨基酸;并且插入是指增加与占据某位置的氨基酸相邻的氨基酸。具体实施方式本发明的第一方面涉及将两个或更多个拷贝的目标多核苷酸整合到真菌宿主细胞的染色体中的方法,该方法通过以下步骤进行:(a)提供在其染色体内包括至少两个整合位点的真菌宿主细胞,每个整合位点包括一对位点特异性重组酶的识别序列,每对识别序列位于驻留(resident)选择标记的两侧;(b)向所述细胞中引入核酸构建体,该核酸构建体包括一对位点特异性重组酶的识别序列,该识别序列对位于目标多核苷酸的两侧;(c)在细胞中短暂表达位点特异性重组酶,借此染色体的识别序列对通过重组酶与核酸构建体的对应的识别序列对重组,从而使得在至少两个整合位点,染色体中的驻留选择标记被切除而目标多核苷酸的拷贝整合到它的位置上,产生包括两个或更多个拷贝的整合到真菌宿主细胞的染色体中的目标多核苷酸的真菌宿主细胞。在一个具体实施方式中,本发明的第一方面涉及将两个或更多个拷贝的目标多核苷酸同时整合到真菌宿主细胞的染色体中的方法,该方法包括以下步骤:(a)提供在其染色体内包括至少两对位点特异性重组酶的识别序列的真菌宿主细胞,每对识别序列位于驻留阴性选择标记的两侧;(b)向所述细胞中引入核酸构建体,该核酸构建体包括一对位点特异性重组酶的识别序列,该识别序列对位于目标多核苷酸的两侧;(c)在细胞中短暂表达位点特异性重组酶,借此染色体的识别序列对通过重组酶与核酸构建体的对应的识别序列对重组,从而使得染色体中的每个驻留阴性选择标记被切除而目标多核苷酸的拷贝整合到它的位置上;然后(d)在选择性培养基中培养该细胞并选择细胞,其中每个阴性选择标记已经通过双同源重组被两个或更多个拷贝的目标多核苷酸取代。在优选的实施方式中,该目标多核苷酸包括编码至少一个目标多肽的操纵子或开放阅读框。该目标多肽可以编码任何目标蛋白质,比如,例如,细胞因子(特别是白介素、干扰素、集落刺激因子(csf)和生长因子),抗凝血剂,酶和酶抑制剂。优选地,该目标多肽包括酶,优选为水解酶、异构酶、连接酶、裂解酶、氧化还原酶或转移酶,例如氨肽酶、淀粉酶、糖酶、羧肽酶、过氧化氢酶、纤维二糖水解酶、纤维素酶、几丁质酶、角质酶、环糊精糖基转移酶、脱氧核糖核酸酶、内切葡聚糖酶、酯酶、α-半乳糖苷酶、β-半乳糖苷酶、葡糖淀粉酶、α-葡糖苷酶、β-葡糖苷酶、转化酶、漆酶、脂肪酶、甘露糖苷酶、变聚糖酶(mutanase)、氧化酶、果胶分解酶、过氧化物酶、植酸酶、多酚氧化酶、蛋白水解酶、核糖核酸酶、转谷氨酰胺酶、木聚糖酶、或β-木糖苷酶。自由可逆的位点特异性重组体系已经在文献中有详细描述。这些可逆体系包括以下:来自噬菌体p1的cre-lox体系(baubonis和sauer,1993,见上文;albert等,1995plantj.,7549-59),酿酒酵母(saccharomycescerevisiae)的flp-frt体系(o'gorrnan等,1991,见上文),zygosaccharonzycesrouxii的r-rs体系(onouchi等,1995mol.gen.genet.247:653-660),来自mu噬菌体的改良gin-gix体系(maeser和kahmann,1991mol.gen.genet.,230:170-76),来自枯草芽孢杆菌(bacillussubtilis)质粒的β-重组酶-six体系(diaz等,1999j.biol.chem.274:6634-6640)以及来自细菌转座子tn1000的δ-γ-res体系(schwikardi和dorge,2000ebslet.471:147-150)。cre、flp、r、gin、β-重组酶和δ-γ是重组酶,lox、frt、rs、gix、six和res是各个重组位点(由sadowslu,1993fasebj.,7:750-67;ow和medberry,1995crit.rev.plantsci.14:239-261综述)。多重cre/lox重组允许选择性位点特异性dna靶向酵母基因组中的自然位点和工程化位点(sauer,b.nucleicacidsresearch.1996,vol.24(23):4608-4613)。已经显示使用链霉菌属(streptomyces)噬菌体φc31的衍生物感染具有自然附着位点attb和异位引入的attb位点的宿主细胞,结果使该噬菌体整合到两个attb位点中(smith等,2004,switchingthepolarityofabacteriophageintegrationsystem.molmicrobiol51(6):1719-1728)。使用mx9噬菌体转化体系,多个拷贝的基因可以引入到包含多个被mx9整合酶识别的附着位点的细胞中(wo2004/018635a2)。温带lactococcal噬菌体tp901-1整合酶和识别序列是良好表征的(breüner等,(1990)novelorganizationofgenesinvolvedinprophageexcisionidentifiedinthetemperatelactococcalbacteriophagetp901-1.jbacteriol181(23):7291-7297;breüner等,2001.resolvase-likerecombinationperformedbythetp901-1integrase.microbiology147:2051-2063)。以上的位点特异性重组体系具有共同的性质,即单一的多肽重组酶催化相同序列或近乎相同序列在两个位点间的重组。每个重组位点由短的不对称间隔序列组成,在该短的不对称间隔序列处发生链交换,该短的不对称间隔序列在两侧有重组酶结合的反向重复序列。间隔序列的不对称性对重组位点提供了一个方向,并规定了重组反应的结果。直接或间接定向的顺式位点之间的重组分别切除或倒置干预dna。反式位点之间的重组引起两个线性dna分子的相互易位,或者,如果两个分子之中有至少一个是环形的,则引起共整合。因为由重组产生的产物位点自身是随后重组的底物,所以反应是自由可逆的。但是,在实际中,因为两个重组位点紧密相连的地方的分子内相互作用的可能性远远高于不相连位点间的分子内相互作用的可能性,所以切除基本上是不可逆的。必然的结果是插入到基因组重组位点中的dna分子会很容易地切除掉,除非该重组酶短暂表达,在这种情况下,插入的dna在重组酶不再表达后得以保持。因此,在第一方面的方法中,优选位点特异性重组酶和它的识别序列对来自于噬菌体p1的cre-lox体系、酿酒酵母的flp-frt体系、zygosaccharonzycesrouxii的r-rs体系、来自mu噬菌体的改良gin-gix体系、来自枯草芽孢杆菌质粒的β-重组酶-six体系,来自细菌转座子tn1000的δ-γ-res体系、链霉菌属噬菌体φc31、mx9噬菌体转化体系或温带lactococcal噬菌体tp901-1的xis-att体系。在实施方式中,位点特异性重组酶和它的识别序列对来自flp-frt体系。在具体的实施方式中,flp重组酶是如在buchholz,frank,improvedpropertiesofflprecombinaseevolvedbycyclingmutagenesis,naturebiotechnologyvolume:16issue:7(1998-07-01)p.657-662中记载的flp重组酶变体。在另一个具体的实施方式中,flp重组酶是具有氨基酸改变p2s、l33s、y108n和s294p的被指定为“flpe”的热稳定性重组酶变体。flpe的核酸序列和相应的氨基酸序列分别见seqidno:106和seqidno:107。在第一方面的优选实施方式中,阴性选择标记编码对宿主细胞赋予抗菌素抗性的多肽且选择性培养基包括抑菌浓度的抗菌素。或者,在第一方面的另一个优选实施方式中,阴性选择标记编码胞嘧啶脱氨酶且选择性培养基包括足量的5-氟胞嘧啶,其通过所述胞嘧啶脱氨酶转化为抑菌浓度的毒性5-氟尿嘧啶。在实施方式中,阴性选择标记编码胞嘧啶脱氨酶多肽,该胞嘧啶脱氨酶多肽与seqidno:60具有至少60%的序列同一性,例如,至少65%,至少70%,至少75%,至少80%,至少85%,至少90%,至少91%,至少92%,至少93%,至少94%,至少95%,至少96%,至少97%,至少98%,至少99%或100%的序列同一性。在一个方面,多肽与seqidno:60相差不多于十个氨基酸,例如九个氨基酸、八个氨基酸、七个氨基酸、六个氨基酸、五个氨基酸、四个氨基酸、三个氨基酸、二个氨基酸或一个氨基酸。优选地,本发明的编码的胞嘧啶脱氨酶多肽包括seqidno:60的氨基酸序列或其等位变体或者由其组成;或者该胞嘧啶脱氨酶多肽是它的具有胞嘧啶脱氨酶活性的片段。在另一个方面,该多肽包括或者由seqidno:60的多肽组成。在另一个实施方式中,阴性选择标记编码胞嘧啶脱氨酶多肽,并在极低等严格条件、低等严格条件、中等严格条件、中高等严格条件、高等严格条件或极高等严格条件下与(i)seqidno:59的多肽编码序列,(ii)其cdna序列,或(iii),(i)或(ii)的全长互补物杂交(sambrook等,1989,molecularcloning,alaboratorymanual,第2版,冷泉港(coldspringharbor),纽约)。根据本领域众所周知的方法,seqidno:59的多核苷酸或其子序列以及seqidno:60的多肽或其片段可以用来设计核酸探针,以鉴定和克隆来自不同属或种的菌株的编码具有胞嘧啶脱氨酶活性的多肽的dna。具体地,根据标准southern印迹步骤,这些探针可以用于与目标细胞的基因组dna或cdna进行杂交,以鉴定并分离其中的相应基因。在长度上,这些探针可以比完整序列短很多,但应当是至少15个核苷酸,例如,在长度上为至少25个,至少35个或至少个70核苷酸。优选地,核酸探针在长度上是至少100个核苷酸,例如,在长度上为至少200个核苷酸,至少300个核苷酸,至少400个核苷酸,至少500个核苷酸,至少600个核苷酸,至少700个核苷酸,至少800个核苷酸或至少900个核苷酸。dna和rna探针两者都可以使用。通常,这些探针被标记以检测相应基因(例如,用32p、3h、35s、生物素或抗生物素蛋白标记)。本发明涵盖这些探针。对于与上述探针杂交且编码具有胞嘧啶脱氨酶活性的多肽的dna,可以筛选从这样的其他菌株制备的基因组dna或cdna文库。可以通过琼脂糖或聚丙烯酰胺凝胶电泳或其它分离技术,分离来自这样的其他菌株的基因组dna或其它dna。来自文库的dna或分离的dna可以被转移到并被固定在硝化纤维或其它合适的载体材料上。为了鉴定与seqidno:59或其子序列同源的克隆或dna,优选在southern印迹中使用载体材料。出于本发明的目的,杂交是指多核苷酸在极低等到极高等严格条件下与如下的标记核酸探针杂交,该标记的核酸探针与(i)seqidno:59;(ii)seqidno:59的多肽编码序列;(iii)其cdna序列;(iv)其全长互补物;或(v)其子序列相对应。可以运用例如x-ray膜来检测核酸探针在这些条件下所杂交的分子。对于长度为至少100个核苷酸的探针,极低等严格条件定义为,在42℃下在5×sspe、0.3%sds、200mg/ml剪切并变性的鲑鱼精子dna和25%甲酰胺中预杂交并杂交,最佳地,之后进行12~24个小时的标准southern印迹过程。载体材料最终使用2×ssc、0.2%sds在45℃下清洗三次,每次15分钟。对于长度为至少100个核苷酸的探针,低等严格条件定义为,在42℃下在5×sspe、0.3%sds、200mg/ml剪切并变性的鲑鱼精子dna和25%甲酰胺中预杂交并杂交,最佳地,之后进行12~24个小时的标准southern印迹过程。载体材料最终使用2×ssc、0.2%sds在50℃下清洗三次,每次15分钟。对于长度为至少100个核苷酸的探针,中等严格条件定义为,在42℃下在5×sspe、0.3%sds、200mg/ml剪切并变性的鲑鱼精子dna和35%甲酰胺中预杂交并杂交,最佳地,之后进行12~24个小时的标准southern印迹过程。载体材料最终使用2×ssc、0.2%sds在55℃下清洗三次,每次15分钟。对于长度为至少100个核苷酸的探针,中高等严格条件定义为,在42℃下在5×sspe、0.3%sds、200mg/ml剪切并变性的鲑鱼精子dna和35%甲酰胺中预杂交并杂交,最佳地,之后进行12~24个小时的标准southern印迹过程。载体材料最终使用2×ssc、0.2%sds在60℃下清洗三次,每次15分钟。对于长度为至少100个核苷酸的探针,高等严格条件定义为,在42℃下在5×sspe、0.3%sds、200mg/ml剪切并变性的鲑鱼精子dna和50%甲酰胺中预杂交并杂交,最佳地,之后进行12~24个小时的标准southern印迹过程。载体材料最终使用2×ssc、0.2%sds在65℃下清洗三次,每次15分钟。对于长度为至少100个核苷酸的探针,极高等严格条件定义为,在42℃下在5×sspe、0.3%sds、200mg/ml剪切并变性的鲑鱼精子dna和50%甲酰胺中预杂交并杂交,最佳地,之后进行12~24个小时的标准southern印迹过程。载体材料最终使用2×ssc、0.2%sds在70℃下清洗三次,每次15分钟。在另一个实施方式中,第一方面的阴性选择标记与seqidno:59的多肽编码序列或其cdna序列具有至少60%的序列同一性,例如,至少65%,至少70%,至少75%,至少80%,至少85%,至少90%,至少91%,至少92%,至少93%,至少94%,至少95%,至少96%,至少97%,至少98%,至少99%或100%的序列同一性。在另一个实施方式中,阴性选择标记编码包括在一个或多个(例如数个)位置的取代、缺失和/或插入的seqidno:60的胞嘧啶脱氨酶多肽的变体。氨基酸变化优选性质小的变化,即不显著影响蛋白质的折叠和/或活性的保守性氨基酸取代或插入;小的缺失,通常为1到大约30个氨基酸的缺失;小的氨基端或羧基端延伸,诸如氨基端的蛋氨酸残基;最多约20~25个残基的小接头肽;或者通过改变净电荷或其它功能而有助于纯化的小延伸,诸如多聚组氨酸片段(tract)、抗原性表位或结合结构域。保守性取代的实例是碱性氨基酸(精氨酸、赖氨酸和组氨酸)、酸性氨基酸(谷氨酸和天冬氨酸)、极性氨基酸(谷氨酰胺和天冬酰胺)、疏水氨基酸(亮氨酸、异亮氨酸和缬氨酸)、芳香族氨基酸(苯丙氨酸、色氨酸和酪氨酸)和小氨基酸(甘氨酸、丙氨酸、丝氨酸、苏氨酸和蛋氨酸)各组内的取代。通常不改变比活度的氨基酸取代在本领域是已知的,并且在例如h.neurathandr.l.hill,1979,in,theproteins,academicpress,newyork中记载。常见的取代是ala/ser、val/ile、asp/glu、thr/ser、ala/gly、ala/thr、ser/asn、ala/val、ser/gly、tyr/phe、ala/pro、lys/arg、asp/asn、leu/ile、leu/val、ala/glu和asp/gly。或者,氨基酸改变具有这样的性质,即多肽的理化性质发生改变。例如,氨基酸改变可以提高多肽的热稳定性,改变底物专一性、改变最适ph等等。多肽中的必需氨基酸可以根据本领域已知的程序如定点诱变或丙氨酸扫描诱变加以鉴定(cunninghamandwells,1989,science244:1081-1085)。在后述技术中,在分子中的每个残基处引入单一丙氨酸突变,且对生成的突变分子测试胞嘧啶脱氨酶活性,以鉴定对该分子的活性很关键的氨基酸残基。还参见hilton等,1996,j.biol.chem.271:4699-4708。酶的活性位点或其他生物相互作用同样可以通过结构的物理分析测定,如通过这样的技术测定,如核磁共振、结晶学、电子衍射或光亲合标记,连同假定接触位点氨基酸的突变。例如,参阅devos等,1992,science255:306-312;smith等,1992,j.mol.biol.224:899-904;wlodaver等,1992,febslett.309:59-64。必需氨基酸的鉴定也可以从与相关多肽的比对来推断。使用已知的诱变、重组和/或改组(shuffling)方法,接着进行相关的筛选步骤,比如reidhaar-olsonandsauer,1988,science241:53-57;bowieandsauer,1989,proc.natl.acad.sci.usa86:2152-2156;wo95/17413;或wo95/22625所公开的,可以完成和测试单个或多个氨基酸的取代、缺失和/或插入。可以运用的其他方法包括易错pcr、噬菌体展示(例如,lowman等,1991,biochemistry30:10832-10837;u.s.专利号5,223,409;wo92/06204)和定位诱变(derbyshire等,1986,gene46:145;ner等,1988,dna7:127)。诱变/改组方法可以与高通量的自动筛选方法结合,以检测宿主细胞所表达的克隆的诱变多肽的活性(ness等,1999,naturebiotechnology17:893-896)。可以从宿主细胞回收编码活性多肽的诱变dna分子且可以用本领域的标准方法来快速测序。这些方法允许对多肽中个别氨基酸残基的重要性进行快速测定。在实施方式中,引入到seqidno:60多肽中的氨基酸取代、缺失和/或插入的数目不多于10,例如,1、2、3、4、5、6、7、8或9。多肽可以是杂合多肽,在其中,一个多肽的区域融合在另一个多肽的区域的n端或c端。具有胞嘧啶脱氨酶活性的多肽的来源编码本发明的具有胞嘧啶脱氨酶活性的多肽的多核苷酸可以从任何属的微生物中获得。出于本发明的目的,本文中与给定来源一起使用的术语“由……获得”应指,由多核苷酸编码的多肽是由该来源或插入了来自该来源的多核苷酸的菌株所产生。胞嘧啶脱氨酶多肽可以是真菌多肽。例如,该多肽可以是酵母多肽,诸如假丝酵母属(candida)、克鲁维酵母属(kluyveromyces)、毕赤酵母属(pichia)、酵母属(saccharomyces)、裂殖酵母属(schizosaccharomyces)或耶氏酵母属(yarrowia)多肽;或丝状真菌多肽,诸如支顶孢属(acremonium)、伞菌属(agaricus)、链格孢属(alternaria)、曲霉属(aspergillus)、短梗霉属(aureobasidium)、botryospaeria、拟蜡菌属(ceriporiopsis)、毛喙壳属(chaetomidium)、金孢子菌属(chrysosporium)、麦角菌属(claviceps)、旋孢腔菌(cochliobolus)、鬼伞属(coprinopsis)、乳白蚁属(coptotermes)、棒囊壳属(corynascus)、栗疫属(cryphonectria)、隐球酵母属(cryptococcus)、色二孢属(diplodia)、黑耳属(exidia)、filibasidium、镰孢霉属(fusarium)、赤霉菌属(gibberella)、全鞭毛虫属(holomastigotoides)、腐质霉属(humicola)、耙齿菌属(irpex)、香菇属(lentinula)、leptospaeria、稻瘟菌属(magnaporthe)、melanocarpus、meripilus、毛霉属(mucor)、毁丝霉属(myceliophthora)、neocallimastix、脉孢菌属(neurospora)、拟青霉属(paecilomyces)、青霉属(penicillium)、平革菌属(phanerochaete)、piromyces、poitrasia、假黑盘菌属(pseudoplectania)、pseudotrichonympha、根毛霉属(rhizomucor)、裂褶菌属(schizophyllum)、柱顶孢属(scytalidium)、踝节菌属(talaromyces)、嗜热子囊菌属(thermoascus)、梭孢壳属(thielavia)、弯颈霉属(tolypocladium)、木霉属(trichoderma)、长毛盘菌属(trichophaea)、轮枝孢属(verticillium)、小包脚菇属(volvariella)或炭角菌属(xylaria)多肽。另一方面,多肽是卡氏酵母(saccharomycescarlsbergensis)、酿酒酵母(saccharomycescerevisiae)、糖化酵母(saccharomycesdiastaticus)、道格拉氏酵母(saccharomycesdouglasii)、克鲁弗酵母(saccharomyceskluyveri)、诺地酵母(saccharomycesnorbensis)或saccharomycesoviformis多肽。另一方面,多肽是解纤维枝顶孢霉(acremoniumcellulolyticus)、棘孢曲霉(aspergillusaculeatus)、泡盛曲霉(aspergillusawamori)、臭曲霉(aspergillusfoetidus)、烟曲霉(aspergillusfumigatus)、日本曲霉(aspergillusjaponicus)、构巢曲霉(aspergillusnidulans)、黑曲霉(aspergillusniger)、米曲霉(aspergillusoryzae)、chrysosporiuminops、嗜角质金孢子菌(chrysosporiumkeratinophilum)、chrysosporiumlucknowense、chrysosporiummerdarium、chrysosporiumpannicola、chrysosporiumqueenslandicum、热带金孢子菌(chrysosporiumtropicum)、chrysosporiumzonatum、杆孢状镰孢(fusariumbactridioides)、fusariumcerealis、克地镰刀菌(fusariumcrookwellense)、黄色镰孢菌(fusariumculmorum)、禾谷镰刀菌(fusariumgraminearum)、禾赤镰孢(fusariumgraminum)、异孢镰孢(fusariumheterosporum)、合欢木镰孢(fusariumnegundi)、尖孢镰孢菌(fusariumoxysporum)、fusariumreticulatum、粉红镰刀菌(fusariumroseum)、接骨木镰孢(fusariumsambucinum)、肤色镰孢(fusariumsarcochroum)、拟枝孢镰刀菌(fusariumsporotrichioides)、硫色镰孢(fusariumsulphureum)、fusariumtorulosum、拟丝孢镰刀菌(fusariumtrichothecioides)、fusariumvenenatum、灰腐质霉(humicolagrisea)、特异腐质霉(humicolainsolens)、柔毛腐质霉(humicolalanuginosa)、白耙齿菌(irpexlacteus)、米赫毛霉(mucormiehei)、嗜热毁丝霉(myceliophthorathermophila)、粗糙脉孢菌(neurosporacrassa)、绳状青霉(penicilliumfuniculosum)、产紫青霉菌(penicilliumpurpurogenum)、黄孢原毛平革菌(phanerochaetechrysosporium)、thielaviaachromatica、thielaviaalbomyces、thielaviaalbopilosa、澳洲梭孢壳(thielaviaaustraleinsis)、thielaviafimeti、小孢梭孢壳(thielaviamicrospora)、卵孢梭孢壳(thielaviaovispora)、thielaviaperuviana、毛梭孢壳(thielaviasetosa)、thielaviaspededonium、耐热梭孢壳(thielaviasubthermophila)、太瑞斯梭孢壳霉(thielaviaterrestris)、哈茨木霉(trichodermaharzianum)、康宁木霉(trichodermakoningii)、长梗木霉(trichodermalongibrachiatum)、里氏木霉(trichodermareesei)或绿色木霉(trichodermaviride)多肽。应当理解,对于前面提及的物种,本发明涵盖上述物种的完全和不完全状态(imperfectstate)和其它分类学同等物,如无性型,而不论这些物种现在的名称如何。本领域技术人员将容易领会适当同等物的具体内容(identity)。这些物种的菌株可以从大量的培养物收藏中心为公众获得,诸如美国典型培养物收藏中心(americantypeculturecollection,atcc)、deutschesammlungvonmikroorganismenundzellkulturengmbh(dsmz)、centraalbureauvoorschimmelcultures(cbs)和北方地区研究中心(nrrl)的农业研究机构专利培养物收藏中心。此外,可以使用上述探针由其它来源包括由自然界(如土壤、堆肥、水等等)分离的微生物鉴定并获得多肽。用于由天然栖息地分离微生物的技术在本领域是众所周知的。然后,相似地,可以通过对另一微生物的基因组dna或cdna文库或者混合的dna样本进行筛选而获得编码多肽的多核苷酸。在用探针检测到编码多肽的多核苷酸后,可以通过本领域普通技术人员已知的技术分离或克隆多核苷酸(参阅sambrook等人,1989,见上文)。核酸构建体本发明还涉及包括选择标记和目标多核苷酸的核酸构建体或表达载体,该选择标记和目标多核苷酸与一个或多个在合适的表达宿主细胞中指导表达的控制序列可操作地连接。在具体的实施方式中,本发明还涉及包括阴性选择标记和目标多核苷酸的核酸构建体或表达载体,该阴性选择标记和目标多核苷酸与一个或多个在合适的表达宿主细胞中指导表达的控制序列可操作地连接。可以通过多种方式操纵多核苷酸以提供所编码的多肽的表达。根据该表达载体,在多核苷酸插入到载体之前对多核苷酸进行操纵是合乎需要的或必须的。使用重组dna方法修饰多核苷酸的技术在本领域是众所周知的。控制序列可以是启动子序列,一种由宿主细胞识别以表达编码本发明多肽的多核苷酸的多核苷酸。启动子序列包含介导多肽表达的转录控制序列。启动子可以是任何在选择的宿主细胞中表现出转录活性的多核苷酸,包括突变、截短和杂合的启动子,并且可从编码胞外或胞内多肽的对于宿主细胞为同源或异源的基因得到。用于在丝状真菌宿主细胞中指导本发明核酸构建体转录的合适启动子的实例是包括从构巢曲霉乙酰胺酶、黑曲霉中性α淀粉酶、黑曲霉酸稳定α淀粉酶、黑曲霉或泡盛曲霉(aspergillusawamori)葡糖淀粉酶(glaa)、米曲霉(aspergillusoryzae)taka淀粉酶、米曲霉碱性蛋白酶、米曲霉磷酸丙糖异构酶、尖孢镰刀菌(fusariumoxysporum)胰酶样蛋白酶(wo96/00787)、fusariumvenenatum淀粉葡糖苷酶(wo00/56900)、fusariumvenenatumdaria(wo00/56900)、fusariumvenenatumquinn(wo00/56900)、米黑根毛霉(rhizomucormiehei)脂肪酶、米黑根毛霉天冬氨酸蛋白酶、里氏木霉(trichodermareesei)β-葡糖苷酶、里氏木霉纤维二糖水解酶i、里氏木霉纤维二糖水解酶ii、里氏木霉内切葡聚糖酶i、里氏木霉内切葡聚糖酶ii、里氏木霉内切葡聚糖酶iii、里氏木霉内切葡聚糖酶iv、里氏木霉内切葡聚糖酶v、里氏木霉木聚糖酶i、里氏木霉木聚糖酶ii、里氏木霉β-木糖苷酶的基因获得的启动子,以及na2-tpi启动子(编码中性α淀粉酶的曲霉基因的修饰启动子,其中不翻译的前导被编码磷酸丙糖异构酶的曲霉基因的不翻译前导替代;非限制性实例包括来自编码中性α淀粉酶的黑曲霉基因的修饰启动子,其中不翻译前导被编码磷酸丙糖异构酶的构巢曲霉或米曲霉基因的不翻译前导替代);及其突变的、截短的和杂合的启动子。在酵母宿主中,有用的启动子从酿酒酵母(saccharomycescerevisiae)烯醇酶(eno-1)、酿酒酵母半乳糖激酶(gal1)、酿酒酵母醇脱氢酶/甘油醛-3-磷酸脱氢酶(adh1、adh2/gap)、酿酒酵母磷酸丙糖异构酶(tpi)、酿酒酵母金属硫蛋白(cup1)以及酿酒酵母3-磷酸甘油酸激酶的基因获得。其它用于酵母宿主细胞的有用启动子在romanos等人(1992,酵母(yeast)8:423-488)中记载。控制序列还可以是合适的转录终止子序列,其由宿主细胞识别以终止转录。终止子序列与编码多肽的多核苷酸的3'末端可操作地连接。在选定的宿主细胞中发挥功能的任何终止子都可以用于本发明。用于丝状真菌宿主细胞的优选终止子从构巢曲霉的邻氨基苯甲酸合成酶基因、黑曲霉的葡糖淀粉酶基因、黑曲霉的α-葡糖苷酶基因、米曲霉的taka淀粉酶基因和尖孢镰刀菌的类胰蛋白酶蛋白酶基因获得。用于酵母宿主细胞的优选终止子从酿酒酵母的烯醇酶基因、酿酒酵母的细胞色素c(cyc1)基因和酿酒酵母的甘油醛-3-磷酸脱氢酶基因获得。其它用于酵母宿主细胞的有用终止子在romanos等人(1992,见上文)中记载。控制序列还可以是合适的前导序列,其在转录时是mrna的非翻译区且对于宿主细胞进行的翻译很重要。前导序列与编码多肽的多核苷酸的5’端可操作地连接。可使用任何在选择的宿主细胞中有功能的前导序列。用于丝状真菌宿主细胞的优选前导序列从米曲霉的taka淀粉酶基因和构巢曲霉的磷酸丙糖异构酶基因获得。用于酵母宿主细胞的合适前导序列由酿酒酵母的烯醇酶(eno-1)基因、酿酒酵母的3-磷酸甘油酸激酶基因、酿酒酵母的α-因子基因和酿酒酵母的醇脱氢酶/甘油醛-3-磷酸脱氢酶(adh2/gap)基因获得。控制序列还可以是多腺苷酸化序列,即与多核苷酸的3'末端可操作地连接、当转录时由宿主细胞识别作为向转录的mrna添加多腺苷酸残基的信号的序列。可以使用在选定的宿主细胞中发挥功能的任何多腺苷酸化序列。用于丝状真菌宿主细胞的优选多腺苷酸化序列由米曲霉的taka淀粉酶基因、黑曲霉的葡糖淀粉酶基因、构巢曲霉的邻氨基苯甲酸合成酶基因、尖孢镰刀菌的类胰蛋白酶蛋白酶基因和黑曲霉的α-葡糖苷酶基因获得。guo和sherman(1995,mol.cellularbiol.15:5983-5990)描述了可用于酵母宿主细胞的多腺苷酸化序列。控制序列也可以是编码与多肽的n端连接并指导多肽进入细胞的分泌通路的信号肽的信号肽编码区。多核苷酸的编码序列的5’端本身可包含在翻译阅读框中天然与编码多肽的编码序列片段相连接的信号肽编码序列。或者,编码序列的5’端可含有对于该编码序列为外源的信号肽编码序列。在编码序列并不天然包含信号肽编码序列的情况下可能需要外源的信号肽编码序列。或者,为增强多肽的分泌,可以简单地用外源信号肽编码序列替换天然信号肽编码序列。然而,可以使用指导所表达多肽进入选定宿主细胞的分泌通路的任何信号肽编码序列。用于丝状真菌宿主细胞的有效信号肽编码序列是获自黑曲霉中性淀粉酶、黑曲霉葡糖淀粉酶、米曲霉taka淀粉酶、特异腐质霉(humicolainsolens)纤维素酶、特异腐质酶内切葡聚糖酶v、柔毛腐质霉(humicolalanuginosa)脂肪酶以及米黑根毛霉(rhizomucormiehei)天冬氨酸蛋白酶的基因的信号肽编码序列。可用于酵母宿主细胞的信号肽获自酿酒酵母α因子和酿酒酵母转化酶的基因。其他可用的信号肽编码序列在romanos等,1992(同上)中有过描述。控制序列也可以是编码位于多肽n端的前肽的前肽编码序列。得到的多肽被称为酶原(proenzyme)或多肽原(或在一些情况下为酶原(zymogen))。多肽原通常为无活性的并能够通过前肽的催化切割或自身催化切割而从多肽原转化为活性多肽。当信号肽和前肽序列在多肽的n端同时存在时,前肽序列的位置紧邻于多肽的n端,且信号肽序列的位置紧邻于前肽序列的n端。也可能期望加入相对于宿主细胞的生长对多肽表达进行调控的调控序列。调控体系的例子是使得基因表达响应于化学或物理刺激(包括调控化合物的存在)而打开或关闭的那些。原核系统中的调控体系包括lac、tac和trp操纵基因体系。在酵母中,可以使用adh2体系或gal1体系。在丝状真菌中,可以使用黑曲霉葡糖淀粉酶启动子、米曲霉takaα-淀粉酶启动子和米曲霉葡糖淀粉酶启动子。调控序列的其他例子是允许基因扩增的那些。在真核系统中,这些调控序列包括,在甲氨蝶呤的存在下扩增的二氢叶酸还原酶基因、以及在重金属下扩增的金属硫蛋白基因。在这些情况中,编码多肽的多核苷酸将与调控序列可操作地连接。表达载体本发明还涉及重组表达载体,包括本发明的多核苷酸、启动子、以及转录和翻译终止信号,以及如本文所述的,一对在目标多核苷酸侧翼的位点特异性重组酶的识别序列、启动子和转录及翻译终止信号。可将多种核苷酸和控制序列连接在一起,产生可包括一个或多个方便的限制性位点的重组表达载体,从而可以在这些位点插入或替代编码多肽的多核苷酸。或者,可以通过将多核苷酸或包括该序列的核酸构建体插入到合适的表达载体中来表达多核苷酸。在建立表达载体时,编码序列位于载体中,使得该编码序列与用于表达的适当控制序列可操作地连接。重组表达载体可以是能够方便地进行重组dna程序并能使多核苷酸表达的任何载体(例如,质粒或病毒)。载体的选择将通常取决于载体和载体将要被引入的宿主细胞之间的相容性。载体可以是线性或闭合的环状质粒。载体可以是自主复制型载体,即作为染色体外实体而存在、其复制独立于染色体复制的载体,例如,质粒、染色体外元件、微型染色体或人工染色体。载体可以包括确保自我复制的任何元件(means)。或者,载体可以是,在被引入宿主细胞后整合到基因组中并与其整合的染色体一起复制的载体。此外,可以使用单个载体或质粒,或一起包含将要引入宿主细胞基因组中的总dna的两个或多个载体或质粒,或转座子。载体优选包含一个或多个允许方便地选择转化细胞、转染细胞、转导细胞等细胞的选择标记。选择标记是其产物例如提供杀生物剂或病毒抗性、重金属抗性、相对营养缺陷型的原养型等的基因。用于酵母宿主细胞的合适标记为,ade2、his3、leu2、lys2、met3、trp1和ura3。用于丝状真菌宿主细胞中的选择标记包括但不限于amds(乙酰胺酶)、argb(鸟氨酸氨甲酰基转移酶)、bar(草胺膦乙酰转移酶,phosphinothricinacetyltransferase)、hph(潮霉素磷酸转移酶)、niad(硝酸还原酶)、pyrg(乳清酸核苷-5'-磷酸脱羧酶)、sc(硫酸腺苷酰转移酶,sulfateadenyltransferase)和trpc(邻氨基苯甲酸合酶)、及其等同物。优选用于曲霉细胞中的是构巢曲霉或米曲霉amds和pyrg基因以及吸水链霉菌(streptomyceshygroscopicus)bar基因。用于连接上述元件以构建本发明的重组表达载体的步骤是本领域技术人员熟知的(参见,例如,sambrook等,1989,见上)。宿主细胞本发明还涉及通过本发明的方法转化的重组宿主细胞。本发明还涉及适合于用包含目标多核苷酸的整合核酸构建体向宿主细胞基因组中进行转化的重组宿主细胞,其中目标多核苷酸的侧翼是与胞嘧啶脱氨酶编码基因同源的区域或与分别在该基因的上游和下游的区域同源的区域,该整合核酸构建体指导通过位点特异性双同源重组进行的染色体整合,借此目标多核苷酸整合到该宿主细胞的基因组中,同时该胞嘧啶脱氨酶编码基因被部分或全部切除并因此失活。驻留胞嘧啶脱氨酶编码基因的成功失活在培养基包括包含5-氟胞嘧啶的培养基中是可以选择的,5-氟胞嘧啶通过胞嘧啶脱氨酶转化成有毒的5-氟尿嘧啶。所以,在这样的转化方法中,胞嘧啶脱氨酶编码基因起阴性选择标记的作用,如在本发明的方法中概述的一样。具有不能测量的胞嘧啶脱氨酶活性的宿主细胞适合于这样的转化方法,在该转化方法中,用包括至少一个可表达的编码胞嘧啶脱氨酶的多核苷酸的核酸构建体转化该宿主细胞,然后在有利于胞嘧啶脱氨酶表达的条件下在包括从头嘧啶合成抑制剂的生长培养基中用作阳性选择标记。优选地,从头嘧啶合成抑制剂是n-膦乙酰基-l-天冬氨酸(pala),其抑制天冬氨酸氨甲酰基转移酶。术语“宿主细胞”包括任意因复制过程中发生的突变而与亲本细胞不同的亲本细胞后代。对于宿主细胞的选择将很大程度取决于编码多肽的基因及其来源。宿主细胞可以是真菌细胞。本文中使用的“真菌”包括子囊菌门(ascomycota)、担子菌门(basidiomycota)、壶菌门(chytridiomycota)和接合菌门(zygomycota)(如在hawksworth等,in,ainsworthandbisby’sdictionaryofthefungi,8thedition,1995,cabinternational,universitypress,cambridge,uk中定义的)以及卵菌门(oomycota)(如在hawksworth等,1995,同上,page171中引用的)和所有有丝分裂的孢子真菌(hawksworth等,1995,同上)。真菌宿主细胞可以是酵母细胞。本文中使用的“酵母”包括产子囊酵母(内孢霉目(endomycetales))、产担孢子(basidiosporogenous)酵母以及属于半知菌类(fungiimperfecti)芽孢纲(blastomycetes)的酵母。因为酵母分类将来可能会变化,出于本发明的目的,酵母应按biologyandactivitiesofyeast(skinner,f.a.,passmore,s.m.,anddavenport,r.r.,eds,soc.app.bacteriol.symposiumseriesno.9,1980)中的描述来定义。酵母宿主细胞可以是假丝酵母属(candida)、汉逊酵母属(hansenula)、克鲁维酵母属(kluyveromyces)、毕赤酵母属(pichia)、酵母属(saccharomyces)、裂殖酵母属(schizosaccharomyces)、或耶氏酵母属(yarrowia)细胞,例如乳酸克鲁维酵母(kluyveromyceslactis)、卡尔酵母(saccharomycescarlsbergensis)、酿酒酵母(saccharomycescerevisiae)、糖化酵母(saccharomycesdiastaticus)、saccharomycesdouglasii、克鲁弗酵母(saccharomyceskluyveri)、saccharomycesnorbensis、saccharomycesoviformis或解脂耶氏酵母(yarrowialipolytica)细胞。真菌宿主细胞可以是丝状真菌细胞。“丝状真菌”包括真菌门(eumycota)和卵菌门亚门的所有丝状类型(如hawksworth等,1995,同上,定义的)。丝状真菌的总体特征为,由几丁质、纤维素、葡聚糖、壳聚糖、甘露聚糖和其他复合多糖构成的菌丝体壁。营养生长是通过菌丝延伸进行的,且碳的分解代谢一般是专性需氧的。相反,酵母例如酿酒酵母的营养生长是通过单细胞菌体的出芽来进行的,且碳的分解代谢可以是发酵的。丝状真菌宿主细胞可以是支顶孢属(acremonium)、曲霉属(aspergillus)、短梗霉属(aureobasidium)、烟管菌属(bjerkandera)、拟蜡菌属(ceriporiopsis)、金孢子菌属(chrysosporium)、鬼伞属(coprinus)、革盖菌属(coriolus)、隐球菌属(cryptococcus)、filibasidium、镰孢霉属(fusarium)、腐质霉属(humicola)、稻瘟菌属(magnaporthe)、毛霉属(mucor)、毁丝霉属(myceliophthora)、neocallimastix、脉孢菌属(neurospora)、拟青霉属(paecilomyces)、青霉属(penicillium)、平革菌属(phanerochaete)、射脉菌属(phlebia)、piromyces、侧耳属(pleurotus)、裂褶菌属(schizophyllum)、踝节菌属(talaromyces)、嗜热子囊菌属(thermoascus)、梭孢壳属(thielavia)、弯颈霉属(tolypocladium)、栓菌属(trametes)或木霉属(trichoderma)细胞。例如,该真菌宿主细胞可以是泡盛曲霉(aspergillusawamori)、臭曲霉(aspergillusfoetidus)、烟曲霉(aspergillusfumigatus)、日本曲霉(aspergillusjaponicus)、构巢曲霉(aspergillusnidulans)、黑曲霉(aspergillusniger)、米曲霉(aspergillusoryzae)、烟管菌(bjerkanderaadusta)、干拟蜡菌(ceriporiopsisaneirina)、ceriporiopsiscaregiea、浅黄拟蜡孔菌(ceriporiopsisgilvescens)、ceriporiopsispannocinta、ceriporiopsisrivulosa、ceriporiopsissubrufa、虫拟蜡菌(ceriporiopsissubvermispora)、chrysosporiuminops、嗜角质金孢子菌(chrysosporiumkeratinophilum)、chrysosporiumlucknowense、chrysosporiummerdarium、chrysosporiumpannicola、chrysosporiumqueenslandicum、热带金孢子菌(chrysosporiumtropicum)、chrysosporiumzonatum、灰盖鬼伞(coprinuscinereus)、毛云芝菌(coriolushirsutus)、fusariumbactridioides、fusariumcerealis、fusariumcrookwellense、黄色镰刀菌(fusariumculmorum)、禾谷镰孢菌(fusariumgraminearum)、禾赤镰孢(fusariumgraminum)、异孢镰孢(fusariumheterosporum)、fusariumnegundi、尖孢镰刀菌(fusariumoxysporum)、fusariumreticulatum、粉红色镰刀菌(fusariumroseum)、接骨木镰刀菌(fusariumsambucinum)、肤色镰孢(fusariumsarcochroum)、拟枝孢镰刀菌(fusariumsporotrichioides)、硫色镰刀菌(fusariumsulphureum)、fusariumtorulosum、拟丝孢镰刀菌(fusariumtrichothecioides)、fusariumvenenatum、特异腐质霉(humicolainsolens)、柔毛腐质霉(humicolalanuginosa)、米赫毛霉(mucormiehei)、嗜热毁丝菌(myceliophthorathermophila)、粗糙脉孢菌(neurosporacrassa)、产紫青霉(penicilliumpurpurogenum)、黄孢原毛平革菌(phanerochaetechrysosporium)、脉射侧菌(phlebiaradiata)、白腐菌(pleurotuseryngii)、太瑞斯梭孢壳霉(thielaviaterrestris)、长绒毛栓菌(trametesvillosa)、变色栓菌(trametesversicolor)、哈茨木霉菌(trichodermaharzianum)、康宁木霉(trichodermakoningii)、长梗木霉(trichodermalongibrachiatum)、里氏木霉(trichodermareesei)、或绿色木霉(trichodermaviride)细胞。可以通过涉及原生质体形成、原生质体转化以及细胞壁再生的方法以本质上已知的方式来转化真菌细胞。用于曲霉和木霉宿主细胞的转化的合适过程在ep238023、yelton等,1984,proc.natl.acad.sci.usa81:1470-1474、和christensen等,1988,bio/technology6:1419-1422中有过描述。用于转化镰孢菌属的适当方法描述于malardier等,1989,gene78:147-156和wo96/00787中。酵母可以使用在beckerandguarente,inabelson,j.n.andsimon,m.i.,editors,guidetoyeastgeneticsandmolecularbiology,methodsinenzymology,volume194,pp182-187,academicpress,inc.,newyork;ito等,1983,j.bacteriol.153:163;andhinnen等,1978,proc.natl.acad.sci.usa75:1920中描述的方法进行转化。胞嘧啶脱氨酶活性的去除或降低本发明还涉及产生亲本细胞的突变体的方法,该方法包括使第一方面的多核苷酸或其编码胞嘧啶脱氨酶的部分灭活、破坏或缺失,结果产生当在同样的条件下培养时与亲本细胞相比产生更少或不产生所编码的胞嘧啶脱氨酶的突变细胞。可以运用本领域中熟知的方法(例如插入、破坏、取代或缺失)通过减少或消除多核苷酸的表达来构建突变细胞。在优选的方面,多核苷酸被失活。例如,待修饰或失活的多核苷酸可以是编码区域或其对于活性而言必需的一部分,或编码区域表达所需的调控元件。这样的调控序列或控制序列的实例可以是启动子序列或它的功能性部分,即,足以影响该多核苷酸的表达的部分。用于可能的修饰的其他控制序列包括但不限于前导序列、多腺苷酸化序列、前肽序列、信号肽序列、转录终止子和转录激活因子。可以通过对亲本细胞进行诱变并选择其中多核苷酸的表达降低或消除的突变细胞,进行多核苷酸的修饰或失活。例如,可以通过使用合适的物理或化学诱变剂、通过使用合适的寡核苷酸或通过对dna序列进行pcr产生的诱变来进行诱变,该诱变可以是特异或随机的。此外,可以通过使用这些诱变剂的任意组合进行诱变。适合于本发明目的的物理或化学诱变剂的实例包括紫外线(uv)照射、羟胺、n-甲基-n'-硝基-n-亚硝基胍(mnng)、o-甲基羟胺、亚硝酸、甲基磺酸乙酯(ems)、亚硫酸氢钠、甲酸和核苷酸类似物。当使用这些试剂时,通常通过在合适条件下在选定诱变剂的存在下培育要突变的亲本细胞,并筛选和/或选择表现出降低的基因表达或没有该基因表达的突变细胞来进行诱变。多核苷酸的修饰或失活可以通过在基因中或其转录或翻译所需的调控元件中插入、取代或缺失一个或多个核苷酸来实现。例如,可以插入或除去核苷酸,从而引入终止密码子、引起起始密码子的除去或引起开放阅读框的改变。根据本领域的已知方法,这样的修饰或失活可以通过定点诱变或pcr产生的诱变来实现。虽然从理论上说,可以在体内进行修饰,即,直接在表达待修饰的多核苷酸的细胞上进行修饰,但优选如下文所示例的在体外进行修饰。消除或降低多核苷酸的表达的方便方法的实例是基于基因取代、基因缺失或基因破坏的技术。例如,在基因破坏方法中,在体外对对应于内源多核苷酸的核酸序列进行诱变以产生缺陷的核酸序列,然后将该序列转化到亲本细胞以产生缺陷型基因。通过同源重组,该缺陷核酸序列取代了内源多核苷酸。可能期望该缺陷型多核苷酸还编码可以用于选择其中多核苷酸已被修饰或破坏的转化子的标记物。在一个方面,多核苷酸被如本文所描述的这些可选标记破坏。本发明还涉及抑制具有胞嘧啶脱氨酶活性的多肽在细胞中表达的方法,其包括对细胞施用双链rna(dsrna)分子或者在细胞中表达双链rna(dsrna)分子,其中该dsrna包含本发明多核苷酸的子序列。在优选的方面,dsrna是长约15、16、17、18、19、20、21、22、23、24、25或更多的双链体(duplex)核苷酸。dsrna优选是小干扰rna(sirna)或微rna(mirna)。在优选的方面,dsrna是用于抑制转录的小干扰rna。在另一优选的方面,dsrna是用于抑制翻译的微rna。本发明还涉及用于抑制细胞中多肽表达的这种双链rna(dsrna)分子,其包含seqidno:59的多肽编码序列的一部分。本发明不局限于任何特定的作用机制,dsrna能进入细胞,并引起相似或相同序列的单链rna(ssrna)包括内源mrna的降解。当细胞暴露于dsrna时,来自同源基因的mrna选择性地通过称为rna干扰(rnai)的过程被降解。本发明的dsrna可以用于基因沉默中。在一个方面,本发明提供使用本发明的dsrnai选择性地降解rna的方法。该方法可以在体外、先体外后体内(exvivo)或在体内实践。在一个方面,该dsrna分子可以用于在细胞、器官或动物中产生功能丧失突变(loss-of-functionmutation)。制备和使用选择性降解rna的dsrna分子的方法在本领域中公知;参见,例如,美国专利no.6,489,127;no.6,506,559;no.6,511,824;和no.6,515,109。本发明进一步涉及亲本细胞的突变细胞,其包括编码胞嘧啶脱氨酶多肽的多核苷酸或其控制序列的破坏或缺失或者编码该多肽的沉默基因,这导致突变细胞与亲本细胞相比产生较少的胞嘧啶脱氨酶或不产生胞嘧啶脱氨酶。胞嘧啶脱氨酶缺陷型突变细胞作为宿主细胞用于转化编码目标原生蛋白和异源蛋白的基因是特别有用的。因此,本发明还涉及产生原生或异源多肽的方法,其包括:(a)在有益于产生多肽的条件下培养突变细胞;和(b)回收所述多肽。术语“异源多肽”是指对于宿主细胞而言不是原生的多肽,例如,原生多肽的突变体。宿主细胞可以包括多于一个拷贝的编码原生或异源多肽的多核苷酸。用于培养和纯化目标产物的方法可以用本领域已知的方法进行。重组酶的短暂表达有众多众所周知的简单方法来实现本发明第一方面中步骤(c)的位点特异性重组酶的短暂表达。首先,有利的是,在引入到细胞中的核酸构建体内包含编码重组酶的多核苷酸,即使使用这种方式,核酸构建体在整合后也可以很容易地从细胞中去除而在染色体中留下剩余的整合目标多核苷酸。一个这样的方法被用于实施例中且也在图1中概括出,其中指示出优选的识别位点对,即frt-f和frt-f3,以及flp重组酶编码基因和双向pyrg标记及双转录终止子(在图1中指示为“term”),其对于之后通过双同源重组切除flp基因和pyrg标记充当同源盒。当然,图1中的术语仅是实例且不是意在限制本发明的范围,它们可以被其他众所周知的标记和识别序列对等替换。在第一方面的优选实施方式中,核酸构建体(两侧同样是该对识别序列)进一步包括:进入选择标记和编码位点特异性重组酶的多核苷酸,其依次在两侧是一对同源盒,它们全部与步骤(c)中的目标多核苷酸一起整合。优选地,该进入选择标记使得可以进行阳性选择或阴性选择或者该进入选择标记是双向的。然后,设想该第一方面的方法包括对步骤(c)中通过目标多核苷酸连同进入选择标记和编码位点特异性重组酶的多核苷酸的双同源重组进行的整合进行阳性选择,其中后两者在两侧是同源盒。进一步地,该方法包括对进入选择标记和编码位点特异性重组酶的多核苷酸的每个整合拷贝的切除进行阴性选择的步骤,其中切除通过其侧翼同源盒之间的双同源重组而进行。在另一个优选实施方式中,第二核酸构建体在步骤(b)中被引入该细胞中,该第二核酸构建体是非复制型或是温度敏感复制型,其包括编码该位点特异性重组酶的多核苷酸和选择标记,并分别通过选择压力或在许可温度下的生长而短暂维持在该细胞内,从而使该位点特异性重组酶可以在步骤(c)中短暂表达,其中该选择标记使得可以进行阳性选择或阴性选择或者其是双向的。在最后的优选实施方式中,步骤(a)中的细胞在其染色体内包括至少一个拷贝的与紧密调节型启动子可操作地连接的编码该位点特异性重组酶的多核苷酸,该启动子可以通过改变生长条件而被启动或关闭,例如通过提供特定的碳源或诱导物,从而可以在步骤(c)中进行该位点特异性重组酶的短暂表达。实施例分子克隆技术记载于sambrook,j.,fritsch,e.f.,maniatis,t.(1989)《分子克隆:实验室手册》(molecularcloning:alaboratorymanual)(第2版),冷泉港实验室,冷泉港,纽约。酶用于dna操作的酶(如限制性内切酶、连接酶等等)可得自newenglandbiolabs,inc.且根据供应商的说明书使用。培养基和试剂使用以下培养基和试剂除非另有说明:用于缓冲剂和底物的化学品是分析等级的商品。cove:342.3g/l蔗糖,20ml/lcove盐溶液,10mm乙酰胺,30g/l诺布尔琼脂(nobleagar)。cove顶层琼脂:342.3g/l蔗糖,20ml/lcove盐溶液,10mm乙酰胺,10g/l低熔点琼脂糖。cove-2:30g/l蔗糖,20ml/lcove盐溶液,10mm乙酰胺,30g/l诺布尔琼脂。cove-n(tf)平板由342.3g蔗糖,20mlcove盐溶液,3gnano3,以及30g诺布尔琼脂和水(每升)构成。cove-n平板由30g蔗糖,20mlcove盐溶液,3gnano3,以及30g诺布尔琼脂和水(每升)构成。cove盐溶液由26gkcl,26gmgso4·7h2o,76gkh2po4以及50mlcove微量金属和水(每升)构成。cove微量金属溶液由0.04gnab4o7·10h2o,0.4gcuso4·5h2o,1.2gfeso4·7h2o,1.0gmnso4·h2o,0.8g中性淀粉酶iimoo2·2h2o,以及10.0gznso4·7h2o和水(每升)构成。cove-n顶层琼脂由342.3g蔗糖,20mlcove盐溶液,3gnano3,以及10g低熔点琼脂糖和水(每升)构成。淀粉葡糖苷酶微量金属溶液由6.8gzncl2·7h2o,2.5gcuso4·5h2o,0.24gnicl2·6h2o,13.9gfeso4·7h2o,13.5gmnso4·h2o以及3g柠檬酸和水(每升)构成。ypg由4g酵母提取物,1gkh2po4,0.5gmgso4·7h2o和15g葡萄糖(ph6.0)和水(每升)构成。stc缓冲液由0.8m山梨醇,25mmtris(ph8)以及25mmcacl2和水(每升)构成。stpc缓冲液由其中含40%peg4000的stc缓冲液构成。mlc由40g葡萄糖,50g大豆粉末,4g柠檬酸(ph5.0)和水(每升)构成。mss由70g蔗糖,100g大豆粉末(ph6.0)和水(每升)构成。mu-1由260g麦芽糖糊精,3gmgso4·7h2o,5gkh2po4,6gk2so4,0.5ml淀粉葡糖苷酶微量金属溶液,以及2g尿素(ph4.5)和水(每升)构成。kcl平板由0.6mkcl,20mlcove盐溶液,3gnano3以及30g诺布尔琼脂和水(每升)构成。5-氟胞嘧啶储液:1000mg5-氟胞嘧啶溶解于1ml0.91nacl溶液中。购买的材料(大肠杆菌,质粒和试剂盒)大肠杆菌dh5-α(toyobo)用于质粒构建和扩增。商业质粒/载体topo克隆试剂盒(invitrogen)和pbluescriptiisk-(stratagene#212206)用于pcr片段的克隆。扩增的质粒由质粒试剂盒(qiagen)回收。用dna连接试剂盒(takara)或t4dna连接酶(boehringermannheim)来完成连接。用expandtmpcr体系(boehringermannheim)来进行聚合酶链反应(pcr)。qiaquicktm凝胶提取试剂盒(qiagen)用于pcr片段纯化和从琼脂糖凝胶提取dna片段。菌株wo2000/039322中描述了米曲霉bech-2。构巢曲霉菌株nrrl1092用作供体株。表达宿主菌株黑曲霉nn059095由novozymes分离且是从土壤中分离的黑曲霉nn049184的派生物。对nn059095进行基因改造以破坏淀粉葡糖苷酶活性的表达。wo2005/070962的实施例11中描述了米曲霉toc1512。质粒专利申请wo2006/069289描述了表达质粒phuda440和来自瓣环栓菌(trametescingulata)的淀粉葡糖苷酶的核苷酸序列。wo07045248的实施例9中描述了质粒pjal574和单纯疱疹病毒(hsv)胸苷激酶基因(tk)、构巢曲霉甘油醛-3-磷酸脱氢酶启动子(pgpd)和构巢曲霉色氨酸合成酶终止子(ttrpc)的核苷酸序列。专利公开wo2005070962描述了表达盒质粒pjal790和中性淀粉酶ii启动子(pna2)的核苷酸序列。专利申请10729.000-us中描述了ja126淀粉酶表达载体。专利wo2001/068864的实施例8中描述了质粒pdv8。实施例10中描述了质粒pjal504。实施例10中描述了质粒pjal504-delta-bglll。专利wo2000/050567a1的实施例1中描述了质粒pjal554。质粒pjal574记载于实施例10中。质粒pjal835记载于实施例10中。质粒pjal955记载于实施例10中。质粒pjal1022记载于实施例10中。质粒pjal1025记载于实施例10中。质粒pjal1027记载于实施例10中。质粒pjal1029记载于实施例10中。质粒pjal1120记载于实施例10中。质粒pjal1123记载于实施例10中。质粒pjal1183记载于实施例10中。质粒pjal1194记载于实施例10中。质粒pjal1202记载于实施例10中。专利wo91/17243中记载了质粒ptoc65。质粒puc19:该构建记载于vieira等,1982,基因(gene)19:259-268。质粒4blunt来自invitrogen曲霉的转化曲霉属菌株的转化可使用用于酵母转化的一般方法实现。本发明的优选步骤描述如下。将黑曲霉宿主菌株接种至补充10mm尿苷的100mlypg培养基中,并在32℃以80rpm温育16小时。收集沉淀,用0.6mkcl洗涤,并重悬于20ml含有终浓度为20mg/ml的市售β-葡聚糖酶产物(glucanextm,novozymesa/s,denmark)的0.6mkcl中。将悬液于32℃在振动下(80rpm)温育直至形成原生质体,然后用stc缓冲液洗涤两次。对原生质体用血细胞计数器计数,在stc:stpc:dmso为8:2:0.1的溶液中重悬并调节至2.5×l07个原生质体/ml的终浓度。将大约4μg的质粒dna添加至100μl的原生质体悬液,轻柔混合,并在冰上温育30分钟。添加1ml的sptc,并将原生质体悬液在37℃温育20分钟。在添加10ml的50℃cove或cove-n顶层琼脂糖之后,将反应物倾至cove或cove-n(tf)琼脂平板上,并将平板在32℃温育5日。其它真菌宿主比如木霉属菌株的转化也可以使用用于真菌转化的一般方法实现。用于葡糖淀粉酶产生的sf培养将所选转化子的孢子接种到100mlmlc培养基中,并在30℃下培育2天。将10mlmlc接种到100mlmu-1培养基中,并在30℃下培育7天。通过离心获得上清液。southern杂交从100mlypg培养基的过夜培养物中收集所选转化子的菌丝体,对其用蒸馏水漂洗,干燥,并于-80℃冷冻。将研磨的菌丝体在65℃下与蛋白酶k和rnasea一起孵育1小时。通过酚/chcl3提取两次,接着进行etoh沉淀来回收基因组dna,并在蒸馏水中重悬。使用pcrdig探针合成试剂盒(rocheappliedscience,indianapolisin)按照制造商的说明书来合成非放射性探针。使用qiaquicktm凝胶提取试剂盒(qiageninc.,valencia,ca)根据制造商的说明书对dig标记的探针进行凝胶提纯。将5微克基因组dna用适当的限制性内切酶完全消化16小时(总体积40μl,4u酶/μldna),并在0.8%的琼脂糖凝胶上跑胶。通过0.2mhcl处理,使dna在凝胶中片段化,变性(0.5mnaoh,1.5mnacl),并中和(1mtris,ph7.5;1.5mnacl),随后于20×ssc中转移至hybondn+膜(amersham)。将dna以uv交联到膜上,并在20mldigeasyhyb(rochediagnosticscorporation,mannheim,germany)中于42℃预杂交1小时。将变性的探针直接加入到digeasyhyb缓冲液中,并在42℃下进行过夜杂交。在杂交后洗涤(于2×ssc中进行两次,室温,5分钟,于0.1×ssc中进行两次,68℃,每次15分钟),接着按照制造商的操作指南,使用dig检测系统和cpd-star(roche)进行化学发光检测。将dig标记的dna分子量标记物ii(roche)用作标准标记物。葡糖淀粉酶活性葡糖淀粉酶活性以淀粉葡糖苷酶单位(agu)进行测量。agu定义为在标准条件下(37℃,ph4.3,底物:麦芽糖23.2mm,缓冲液:醋酸盐0.1m,反应时间5分钟)每分钟水解1微摩尔麦芽糖的酶的量。可以使用自动分析仪系统。将变旋酶加至葡萄糖脱氢酶试剂中,从而使得存在的任何α-d-葡萄糖均变为β-d-葡萄糖。在上述反应中葡萄糖脱氢酶特异性地与β-d-葡萄糖反应,形成nadh,nadh使用光度计在340nm下测定,作为原始葡萄糖浓度的测量结果。淀粉葡糖苷酶孵育:显色反应:酸性α-淀粉酶活性的测定当根据本发明使用时,任何酸性α-淀粉酶的活性均可以用afau(酸性真菌α-淀粉酶单位)进行测量,其相对于酶标准物而测定。1fau定义为在下述标准条件下每小时降解5.260mg淀粉干物质的酶的量。酸性α-淀粉酶(即酸稳定的α-淀粉酶),内切-α-淀粉酶(1,4-α-d-葡聚糖-葡聚糖-水解酶,e.c.3.2.1.1)水解淀粉分子内部区域中的α-1,4-糖苷键,形成具有不同链长的糊精和寡糖。与碘形成的颜色强度与淀粉的浓度直接成比例。使用反向比色法将淀粉酶活性测定为指定分析条件下的淀粉浓度的减少。λ=590nm蓝/紫t=23秒脱色标准条件/反应条件:实施例1.在黑曲霉nn059095的中性淀粉酶i(nai)基因座处引入frt位点构建潮霉素b抗性基因表达质粒phuda966基于genbank(id#ab007770)中的核苷酸序列信息,设计分别引入ecori/spei和bamhi位点的以下引物tef-f和tef-r,以分离米曲霉tef1的启动子区(翻译延长因子1/ptef1):tef-f(seqidno:1):gaattcactagtggggttcaaatgcaaacaatef-r(seqidno:2):ggatcctggtgcgaactttgtagtt使用引物对tef-f和tef-r,以米曲霉菌株bech2的基因组dna作为模板,进行pcr反应。反应产物在1.0%琼脂糖凝胶上分离,从胶上切除0.7kb的产物条带。将0.7kb扩增的dna片段用bamhi和ecori消化,并连接到用bamhi和ecori消化的曲霉表达盒phuda440中,产生phuda440-ptef。基于embl:d49701中的核苷酸序列信息,设计分别引入xhoi和xbai位点的以下引物nia-f和nia-r,以分离米曲霉硝酸还原酶(niad)的终止子区(tniad):nia-f(seqidno:3):ctcgagattatccaagggaatgacnia-r(seqidno:4):tctagaaagtattttcggtacgatt使用引物对nia-f和nia-r,以米曲霉菌株bech2的基因组dna作为模板,进行pcr反应。反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除0.5kb的产物条带。将0.5kb扩增的dna片段用xhoi和xbai消化,并连接到用xhoi和xbai消化的曲霉表达盒phuda440-ptef中,以产生phuda440-ptef-tnia。基于embl:ar109978中的核苷酸序列信息,设计分别引入bamh和xhoi位点的以下引物hph-f和hph-r,以分离潮霉素b抗性基因的编码区:hph-f(seqidno:5):ggatcctacacctcagcaatgtcgcctgaahph-r(seqidno:6):ctcgagctattcctttgccctcggacgagtgct使用引物对hph-f和hph-r,以带有潮霉素b抗性基因(hph)的pjal154作为模板,进行pcr反应。反应产物在1.0%琼脂糖凝胶上分离,从凝胶上切除1.0kb的产物条带。将1.0kb的扩增dna片段用bamhi和xhoi消化,并连接到用bamhi和xhoi消化的曲霉表达盒phuda440-ptef-tnia中,以产生phuda966。phuda966中潮霉素b抗性基因(hph)表达部分的核苷酸序列显示在seqidno:7中,其指示出用于进行构建的引物的特征位置,所编码的潮霉素b抗性因子显示于seqidno:8。构建用于在na1基因座处引入frt位点的phuda981通过xhoi和ecori消化,从pjal574中回收含有单纯疱疹病毒(hsv)胸苷激酶基因(tk)的2.5kbdna片段。将回收的2.5kb片段连接到经xhoi和ecori消化的pbluescriptiisk-。将连接混合物转化到大肠杆菌dh5α中,以产生表达质粒ptk。frt-f和frt-f3位点的核苷酸序列为:frt-f(seqidno:9):ttgaagttcctattccgagttcctattctctagaaagtataggaacttcfrt-f3(seqidno:10):ttgaagttcctattccgagttcctattcttcaaatagtataggaacttca分别基于embl:am270106和embl:dj052242中的核苷酸序列信息,设计分别引入ecori和spei位点的以下引物3na1-f和3na1-r,分别,以分离融合有frt-f3识别位点的黑曲霉中性淀粉酶i(nai)的3’侧翼区:3na1-f(seqidno:11):actagtttgaagttcctattccgagttcctattcttcaaatagtataggaacttcaactagagtatatgatggtact3na1-r(seqidno:12):gaattcgcattctcctagttactgatgacttt使用引物对3na1-f和3na1-r,以黑曲霉nn059095的基因组dna作为模板,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除1.0kb的产物条带。将1.5kb的扩增dna片段用spei和ecori消化,并连接到用ecori和spei消化的曲霉表达盒ptk中,以产生phudatk-3na1。分别基于embl:am270106和embl:dj052242中的核苷酸序列信息,设计分别引入noti和spei位点的以下引物5na1-f和5na1-r,以分离融合有frt-f识别位点的黑曲霉中性淀粉酶i(nai)的5’侧翼区:5na1-f(seqidno:13):gcggccgcgtttaaacctatctgttccc5na1-r(seqidno:14):actagtgctagcgaagttcctatactttctagagaataggaactcggaataggaacttcaagatgaattcgcggcctacatg使用引物对5na1-f和5na1-r,以黑曲霉nn059095的基因组dna作为模板,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除1.8kb的产物条带。将1.8kb的扩增dna片段用noti和spei消化,并连接到用noti和spei消化的曲霉表达盒ptk-3na1中,以产生phudatk-3na1-5na1。通过xbai和nhei消化,从phuda966中回收含有由米曲霉tefl启动子(ptef)和niad终止子(tniad)驱动的潮霉素b抗性基因的2.2kbdna片段。将回收的2.2kb片段连接到spei消化的phudatk-3na1-5na1。将连接混合物转化到大肠杆菌dh5α中,以产生表达质粒phuda981。phuda981的na1编码部分和侧翼区的核苷酸序列显示于seqidno:15,na1显示于seqidno:16,且质粒图谱显示于图2。在黑曲霉nn059095的na1基因座处引入frt位点将phuda981引入到黑曲霉菌株nn059095中。从补充有10mm尿苷和1mm潮霉素b的cove-n(tf)中选择转化子。将随机选择的转化子接种到具有10mm尿苷、1mm潮霉素b和2.5μm5-氟-2-脱氧尿苷(fdu)(一种杀死对在phuda981中携带的单纯疱疹病毒(hsv)胸苷激酶基因(tk)进行表达的细胞的试剂)的cove-n平板上。对在补充有2.5μmfdu的cove-n平板上生长良好的菌株进行纯化,并进行southern印迹分析以证实phuda981中的frt位点是否正确引入。使用以下用于非放射性探针的引物组来分析所选择的转化子。对于5’na1侧翼区:正向引物(seqidno:17):aatccggatcctttcctata反向引物(seqidno:18):gatggagcgcgcctagaagc对从所选择的转化子中提取的基因组dna用ncoi消化,并使用上述探针进行southern印迹分析。通过2.8kbncoi条带的消失和3.1kbncoi条带的出现来识别目标菌株。在这些给出正确整合事件的菌株当中,选择出称作nn059180的菌株。实施例2.在黑曲霉nn059095的酸稳定性淀粉酶基因座处引入frt位点构建构巢曲霉乙酰胺酶基因(amds)表达质粒phuda976.基于embl:af348620中的核苷酸序列信息,设计分别引入bamhi和xhoi位点的以下引物amds-f和amds-r,以分离amds基因的编码区:amds-f(seqidno:19):ggatccaccatgcctcaatcctggamds-r(seqidno:20):ctcgagctatggagtcaccacatttcccag使用引物对amds-f和amds-r,以构巢曲霉菌株nrrl1092的基因组dna作为模板,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除1.0kb的产物条带。将1.9kb的扩增dna片段用bamhi和xhoi消化,并连接到用bamhi和xhoi消化的曲霉表达盒phuda440-ptef-tnia中,以产生phuda976。phuda976中构巢曲霉乙酰胺酶基因(amds)表达部分的核苷酸序列显示于seqidno:21,具有所使用的引物的基因特征位置,所编码的乙酰胺酶氨基酸序列显示于seqidno:22。构建用于在酸稳定性淀粉酶基因座处引入frt位点的phuda1019分别基于embl:am270232和embl:dj052242的核苷酸序列信息,设计分别引入ecori和spei位点的以下引物3sp-f和3sp-r,以分离融合有frt-f3识别位点的黑曲霉酸稳定性淀粉酶的3’侧翼区:3sp-f(seqidno:23):actagtttgaagttcctattccgagttcctattcttcaaatagtataggaacttcaactagagaatgcaatcataacagaaagta3sp-r(seqidno:24):gaattcttaattaaatcacggcaagggtttac使用引物对3sp-f和3sp-r,以黑曲霉nn059095的基因组dna作为模板,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除1.8kb的产物条带。将1.8kb的扩增dna片段用spei和ecori消化,并连接到用ecori和spei消化的曲霉表达盒ptk,以产生phudatk-3sp。分别基于embl:am270232和embl:dj052242中的核苷酸序列信息,设计分别引入sacii和spei位点的以下引物5sp-f和5sp-r,以分离融合有frt-f识别位点的黑曲霉酸稳定性淀粉酶的5’侧翼区:5sp-f(seqidno:25):ccgcggcaacaggcagaatatcttcc5sp-r(seqidno:26):actagtgaagttcctatactttctagagaataggaactcggaataggaacttcaaacgggatcttggacgcattcca使用引物对5sp-f和5sp-r,以黑曲霉nn059095的基因组dna作为模板,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除2.0kb的产物条带。将2.0kb的扩增dna片段用sacii和spei消化,并连接到用sacii和spei消化的曲霉表达盒ptk-3sp,以产生phudatk-3sp-5sp。通过xbai和nhei消化,从phuda976中回收含有由米曲霉tefl启动子和niad终止子驱动的amds基因的3.1kbdna片段。将回收的3.1kb片段连接到spei消化的phudatk-3sp-5sp。将连接混合物转化到大肠杆菌dh5α中,以产生表达质粒phuda1019。具有phuda1019侧翼序列的黑曲霉酸稳定性淀粉酶基因的核苷酸序列显示于seqidno:27,所编码的淀粉酶氨基酸序列显示于seqidno:28;质粒图谱显示在图3中。在黑曲霉nn059180中的基因座处引入frt位点将phuda1019引入到黑曲霉菌株nn059180中。从补充有10mm尿苷的cove(tf)中选择转化子。将随机选择的转化子接种到具有10mm尿苷和2.5μm5-氟-2-脱氧尿苷(fdu)(一种杀死对在phuda1019中携带的单纯疱疹病毒(hsv)胸苷激酶基因(tk)进行表达的细胞的试剂)的cove-2平板上。将在具有2.5μmfdu的cove-2平板上生长良好的菌株进行纯化,并进行southern印迹分析,以确认phuda1019中的frt位点是否正确引入。使用用于非放射性探针的以下引物组来分析所选择的转化子。对于5’酸稳定性淀粉酶侧翼区:正向引物(seqidno:29):cgtacaccttgggattatgcgctg反向引物(seqidno:30):cacaaaggcgcaaagcataccatc将从所选择的转化子中提取的基因组dna用xhoi消化。通过6.2kbxhoi条带的消失和4.1kbxhoi条带的出现来识别正确的整合事件。在这些具有正确整合结果的菌株中,选择称作nn059183的菌株。实施例3.在两个基因座中通过flp进行同时位点特异性整合构建构巢曲霉pyrg基因表达质粒phuda794基于embl:m19132中的核苷酸序列信息,设计以下引入paci位点的引物pyr-f以及pyr-r,以分离构巢曲霉pyrg基因的启动子和编码区:pyr-f(seqidno:31):ttaattaaactaaatgacgtttgtgaacapyr-r(seqidno:32):ctaccgccaggtgtcagtcaccctcaaagtccaactcttttc基于embl:am270061和dj052242中的核苷酸序列信息,设计引入sphi位点的以下引物tamg-f和tamg-r,以分离融合有frt-f3识别位点的黑曲霉淀粉葡糖苷酶(tamg)基因的终止子区域:tamg-f(seqidno:33):agagttggactttgagggtgactgacacctggcggtagtamg-r(seqidno:34):gcatgcactagctagttgaagttcctatactatttgaagaataggaactcggaataggaacttcaacctagaggagagagttg使用引物对pyr-f和pyr-r,以构巢曲霉菌株nrrl1092的基因组dna作为模板,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除1.4kb的产物条带。使用引物对tamg-f和tamg-r,以黑曲霉nn059095的基因组dna作为模板,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除0.8kb的产物条带。使用引物对pyr-f和tamg-r,对1.4kb和0.8kb的扩增dna片段进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除2.2kb的产物条带。使用试剂盒,按照操作指南,将2.2kb的扩增dna片段包装到invitrogen提供的topo克隆载体(pcr2.1topo)中,以产生phuda794。phuda794中具有侧翼序列的构巢曲霉pyrg基因的核苷酸序列,连同所用引物的特征和位置显示于seqidno:35;所编码的pyrg的氨基酸序列显示于seqidno:36。构建flp基因合成版本的表达质粒phuda996基于embl:z49892中的核苷酸序列信息,分别设计引入sphi位点和bamhi位点的以下引物xln-f和xln-r,来分离构巢曲霉xlna基因的启动子区(pxlna):xln-f(seqidno:37):gcatgcttaattaatggaagtgcgttgatcattxln-r(seqidno:38):ggatcccctgtcagttggg使用引物对xln-f和xln-r,以构巢曲霉菌株nrrl1092的基因组dna作为模板,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除0.7kb的产物条带。将0.7kb的扩增dna片段用bamhi和sphi消化,并连接到用bamhi和sphi消化的曲霉表达盒phuda966中,以产生phuda966-pxlna。通过bamhi和xhoi消化,从pjal1008中回收含有flp基因合成版本(sflp)的1.3kbdna片段。将回收的1.3kb片段连接到bamhi和xhoi消化的phuda966-pxlna。将连接混合物转化到大肠杆菌dh5α中,以产生表达质粒phuda996。phuda996中flp表达部分的合成版本的核苷酸序列,与所用引物的特征和位置一起显示于seqidno:39;所编码的sflp的氨基酸序列显示于seqidno:40。构建用于在nn059183的中性淀粉酶1(na1)和酸稳定性淀粉酶基因座处同时进行位点特异性整合的phuda1000基于pjal790和embl:dj052242中的核苷酸序列信息,设计分别引入ecori位点和bamhi位点的以下引物pna-f和pna-r,以分离融合有frt-f识别位点的三重串联的黑曲霉中性淀粉酶ii(na2)基因的启动子区(pna2):pna-f(seqidno:41):gaattcatcttgaagttcctattccgagttcctattctctagaaagtataggaacttcgctagccgagagcagcttgaagapna-r(seqidno:42):ggatcccccagttgtgtatatagaggatt使用引物对pna-f和pna-r,以pjal790作为模板,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除1.7kb的产物条带。将1.7kb的扩增dna片段用ecori和bamhi消化,并连接到用ecori和bamhi消化的携带有来自瓣环栓菌的淀粉葡糖苷酶基因(t.c.ga)的曲霉表达盒phuda440,以产生phuda440-frt。通过paci和sphi消化,从phuda794中回收含有构巢曲霉pyrg基因的2.2kbdna片段。将回收的2.2kb片段连接到paci和sphi消化的phuda440-frt。将连接混合物转化到大肠杆菌dh5α中,以产生表达质粒phuda440-frt-pyrg。通过paci和xbai消化,从phuda996中回收含有由xlna启动子和niad终止子驱动的flp基因的2.4kbdna片段。将回收的2.4kb片段连接到paci和xbai消化的phuda440-frt-pyrg。将连接混合物转化到大肠杆菌dh5α中,以产生表达质粒phuda1000。质粒图谱显示于图4。通过flp进行的同时位点特异性整合将phuda1000引入到黑曲霉菌株nn059183中。从补充有1%d-木糖的cove-n(tf)中选择转化子。将随机选择的转化子接种到cove-n平板上。将在cove-n平板上生长良好的菌株进行纯化,并进行southern印迹分析,以确认phuda1000中的表达部分是否正确引入。使用用于非放射性探针的以下引物组来分析所选择的转化子。对于t.c.ga编码区:正向引物(seqidno:43):tcgagtgcggccgacgcgtacgtc反向引物(seqidno:44):cagagagtgttggtcacgta将从所选择的转化子中提取的基因组dna用hindiii消化,并使用上述探针进行southern印迹分析。通过2.8kbncoi条带的消失和3.1kbncoi条带的出现来识别目标菌株。通过正确的整合事件,观察到分别在na1和酸稳定性淀粉酶基因座处引入的7.2kb和5.7kb大小的两个杂交信号。图5示出当phuda1000正确引入到nn059183中时的示意性na1(上图)和酸稳定性淀粉基因座(下图)。实施例4.nn059183中黑曲霉ku70基因的破坏构建黑曲霉ku70基因破坏载体phuda801基于embl:am270339的核苷酸序列信息,设计分别引入ecori位点和spei位点的以下引物3ku-f和3ku-r,以分离黑曲霉ku70基因的3’侧翼区:3ku-f(seqidno:45):actagttctagaagccgtgggtatttttatgaa3ku-r(seqidno:46):gaattcgtttaaacttggcggctgccaagcttcc使用引物对3ku-f和3ku-r,以黑曲霉菌株nn059183的基因组dna作为模板,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除2.0kb的产物条带。将2.0kb的扩增dna片段用ecori和spei消化,并连接到用ecori和spei消化的ptk中,以产生ptk-3ku。基于embl:am270339的核苷酸序列信息,设计分别引入noti位点和spei位点的以下引物5ku-f和5ku-r,以分离黑曲霉ku70基因的5’侧翼区:5ku-f(seqidno:47):gcggccgctcattcagagagctacccgt5ku-r(seqidno:48):actagttaattaagaggaccgcatctttga使用引物对5ku-f和5ku-r,以黑曲霉菌株nn059183的基因组dna作为模板,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除1.3kb的产物条带。将1.3kb的扩增dna片段用noti和spei消化,并连接到用noti和spei消化的ptk-3ku中,以产生ptk-3ku-5ku。通过spei和xbai消化,从phuda794中回收含有构巢曲霉pyrg基因的2.2kbdna片段。将回收的2.2kb片段连接到spei和xbai消化的ptk-3ku-5ku。将连接混合物转化到大肠杆菌dh5α中,以产生表达质粒phuda801。phuda801中黑曲霉ku70基因和侧翼序列的核苷酸序列显示于seqidno:49;ku70所编码多肽的氨基酸序列显示于seqidno:50;质粒图谱显示于图6。nn059183中ku70基因破坏将phuda801引入到黑曲霉菌株nn059183中。从cove-n(tf)中选择转化子。将随机选择的转化子接种到具有2.5μm5-氟-2-脱氧尿苷(fdu)(一种杀死对在phuda801中携带的单纯疱疹病毒(hsv)胸苷激酶基因(tk)进行表达的细胞的试剂)的cove-n平板上。将在具有2.5μmfdu的cove-n平板上生长良好的菌株进行纯化,并进行southern印迹分析,以确认ku70基因是否正确地破坏。使用用于非放射性探针的以下引物组来分析所选择的转化子。对于3’ku70侧翼区:正向引物(seqidno:51):acggtatgcgtacaatgatca反向引物(seqidno:52):atttgagggcaccagcacccc将从所选择的转化子中提取的基因组dna用spei消化。通过正确的基因破坏事件,经spei消化的8.3kb大小的杂交信号变换至被上述探针探测的5.1kb。在这些具有正确整合事件的菌株中,选择称作c1997的菌株。实施例5.在c1997中的两个基因座处通过flp进行同时位点特异性整合c1997中的pyrg基因拯救首先,如下对c1997中ku70基因座处引入的pyrg基因进行拯救。将菌株c1997接种一次到含有10mm尿苷和1g/l5-氟-乳清酸(5-foa)的cove-n培养基中。在5-foa的存在下,pyrg基因已经缺失的菌株将会生长;那些保留该基因的菌株会将5-foa转化为有毒中间体5-氟-ump。分离出生长更加迅速的菌落。所分离的菌株命名为m1117。在m1117中通过flp进行的同时位点特异性整合将phuda1000引入到黑曲霉菌株m1117中。从补充有1g/ld-木糖的cove-n(tf)中选择转化子。将随机选择的转化子接种到cove-n平板上。将在cove-n平板上生长良好的菌株进行纯化,并进行southern印迹分析,以证实phuda1000中的表达部分是否正确引入。使用用于非放射性探针的以下引物组来分析所选择的转化子。对于t.c.ga编码区:正向引物(seqidno:53):tcgagtgcggccgacgcgtacgtc反向引物(seqidno:54):cagagagtgttggtcacgta将从所选择的转化子中提取的基因组dna用hindiii消化。通过正确的整合事件,观察到分别在na1和酸稳定性淀粉酶基因座处引入的7.2kb和5.7kb大小的两个杂交信号。ku70基因破坏(m1117)的同时整合频率为约20%,而ku70基因没有破坏(nn059183)的为约4~5%。这表明,ku70基因破坏在改善经由flp的基因座特异性整合频率中发挥重要作用。实施例6.nn059183中黑曲霉fcy1基因破坏构建黑曲霉(胞嘧啶脱氨酶)fcy1基因破坏载体phuda1043基于embl:am269962中的核苷酸序列信息,设计分别引入xbai位点和pmei位点的以下引物3fcy-f和3fcy-r,以分离黑曲霉fcy1基因的3’侧翼区:3fcy-f(seqidno:55):tctagaattgaaagctagttctggtcgcat3fcy-r(seqidno:56):gtttaaactccttgcttcgcatacatgcccac以黑曲霉菌株nn059183的基因组dna作为模板,使用引物对3fcy-f和3fcy-r,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除2.0kb的产物条带。将2.0kb的扩增dna片段用xbai和pmei消化,并连接到用xbai和pmei消化的phuda801中,以创建phuda801-3fcy。基于embl:am269962中的核苷酸序列信息,设计分别引入noti位点和spei位点的以下引物5fcy-f和5fcy-r,以分离黑曲霉fcy1基因的5’侧翼区:5fcy-f(seqidno:57):gcggccgccgccgccgaagaactgagcaaa5fcy-r(seqidno:58):actagtatatcttcttatcgcagagattg以黑曲霉菌株nn059183的基因组dna作为模板,使用引物对5fcy-f和5fcy-r,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除2.1kb的产物条带。将2.1kb的扩增dna片段用noti和spei消化,并连接到用noti和spei消化的phuda801-3fcy中,以创建phuda1043。phuda1043中黑曲霉fcy1基因的核苷酸序列和侧翼序列显示于seqidno:59;fcy1所编码多肽的氨基酸序列显示于seqidno:60。质粒图谱显示于图7。nn059183中fcy1基因破坏将phuda1043引入到黑曲霉菌株nn059183中。从cove-n(tf)中选择转化子。将随机选择的转化子接种到具有2.5μmfdu(一种杀死对在phuda1043中携带的单纯疱疹病毒(hsv)胸苷激酶基因(tk)进行表达的细胞的试剂)的cove-n平板上。将在具有2.5μmfdu的cove-n平板和具有10μg/ml5-氟胞嘧啶(5fc)的cove-n平板上生长良好的菌株进行纯化,并进行southern印迹分析,以确认fcy1基因是否正确地被破坏。使用用于非放射性探针的以下引物组来分析所选择的转化子。对于3’fcy1侧翼区:正向引物(seqidno:61):gaaagctagttctggtcgcattgagc反向引物(seqidno:62):gaagttgaaggagatgggtctgga将从所选择的转化子中提取的基因组dna用nhei和xhoi消化,并使用上述探针进行southern印迹分析。通过3.1kbnhei-xhoi条带的消失和2.0kbnhei-xhoi条带的出现来识别目标菌株。在这些具有正确整合事件的菌株中,选择菌株nn059186。实施例7.在黑曲霉nn059186的中性淀粉酶ii(na2)基因座处引入frt位点和黑曲霉fcy1基因nn059186中pyrg基因拯救首先,如下对nn059186中在fcy1基因座处引入的pyrg基因进行拯救。将菌株nn059186接种一次到含有10mm尿苷和1g/l5-氟-乳清酸(5-foa)的cove-n培养基中。在5-foa的存在下,pyrg基因已经缺失的菌株将会生长;那些保留该基因的菌株会将5-foa转化为毒性中间体5-氟-ump。分离出生长更加迅速的菌落。所分离的菌株命名为nn059200。构建用于在na2基因座处引入frt位点和黑曲霉fcy1的phuda1078基于embl:am270278和dj052242中的核苷酸序列信息,设计分别引入xbai位点和pmei位点的以下引物3na2-f和3na2-r,以分离融合有frt-f3位点的黑曲霉na2基因的3’侧翼区:3na2-f(seqidno:63):tctagattgaagttcctattccgagttcctattcttcaaatagtataggaacttcatgtctccatgtttcttgagcggaagtact3na2-r(seqidno:64):gtttaaacgaagactgatattatggcggaa以黑曲霉菌株nn059183的基因组dna作为模板,使用引物对3na2-f和3na2-r,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除2.1kb的产物条带。将2.1kb的扩增dna片段用xbai和pmei消化,并连接到用xbai和pmei消化的phuda801,以创建phuda801-3na2。基于embl:am270278和dj052242中的核苷酸序列信息,设计分别引入noti位点和spei位点的以下引物5na2-f和5na2-r,以分离融合有frt-f位点的黑曲霉na2基因的5’侧翼区:5na2-f(seqidno:65):gcggccgcaagagtcaaaagatagcagagc5na2-r(seqidno:66):actagtgctagcgaagttcctatacttgaataggaactcggaataggaacttcaagatgaattcgcggccggccgcatg以黑曲霉菌株nn059183的基因组dna作为模板,使用引物对5na2-f和5na2-r,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除2.0kb的产物条带。将2.0kb的扩增dna片段用noti和spei消化,并连接到用noti和spei消化的phuda801-3na2中,以创建phuda801-3na2-5na2。通过nhei和xbai消化,从phuda440-frt中回收含有由三重串联na2启动子(pna2)和amg终止子(tamg)驱动的t.c.ga基因的4.3kbdna片段。将回收的4.3kb片段连接到nhei和xbai消化的phuda801-3na2-5na2。将连接混合物转化到大肠杆菌dh5α中,以创建表达质粒phuda801-3na2-5na2-tc。通过speii和xbai消化,从phuda794中回收含有构巢曲霉pyrg基因的2.1kbdna片段。将回收的2.1kb片段连接到xbai部分消化的phuda801-3na2-5na2-tc。将连接混合物转化到大肠杆菌dh5α中,以创建表达质粒phuda801-3na2-5na2-tc-pyrg。基于embl:am269962中的核苷酸序列信息,设计在两个位点处均引入nhei位点的以下引物fcy-f和fcy-r,以分离黑曲霉fcy1基因的整个区域:fcy-f(seqidno:67):gctagcgcgaggctatcacggaggctgtggfcy-r(seqidno:68):gctagcttctgtggttcttgccatgatcgt以黑曲霉菌株nn059183的基因组dna作为模板,使用引物对fcy-f和fcy-r,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除1.5kb的产物条带。将1.5kb的扩增dna片段用nhei消化,并连接到用nhei消化的phuda801-3na2-5na2-tc-pyrg,以创建phuda1078。phuda1078中黑曲霉na2基因及侧翼序列的核苷酸序列显示于seqidno:69;na2所编码多肽的氨基酸序列显示于seqidno:70。phuda1078&1067(见下)中黑曲霉fcy1的核苷酸序列显示于seqidno:71,且fcy1编码的氨基酸序列显示于seqidno:72。phuda1078的质粒图谱显示于图8。在黑曲霉nn059200的na2基因座处引入frt位点和黑曲霉fcy1基因及t.c.ga将phuda1078引入到黑曲霉菌株nn059200中。从cove-n(tf)中选择转化子。将随机选择的转化子接种到具有2.5μm5-氟-2-脱氧尿苷(fdu)的cove-n平板上。将在具有2.5μmfdu的cove-n平板上生长良好并在具有10μg/ml5-氟胞嘧啶(5fc)的cove-n平板上几乎不生长的菌株进行纯化,并进行southern印迹分析,以确认frt位点和fcy1/t.c.ga基因是否正确引入在na2基因座上。使用用于非放射性探针的以下引物组来分析所选择的转化子。对于t.c.ga编码区:正向引物(seqidno:73):tcgagtgcggccgacgcgtacgtc反向引物(seqidno:74):cagagagtgttggtcacgta将从所选择的转化子中提取的基因组dna用spei消化。通过正确的基因引入事件,观察到如上所述被探针探测到的spei消化的4.4kb大小的杂交信号。在具有正确整合事件的菌株中,选择菌株nn059203。实施例8.在黑曲霉nn059203的中性淀粉酶i(na1)和酸稳定性淀粉酶基因座处引入frt位点和黑曲霉fcy1基因以及t.c.ga基因nn059203中pyrg基因拯救如下对nn059203中na2基因座处引入的pyrg基因进行拯救。将菌株nn059203接种一次到含有10mm尿苷和1g/l5-氟-乳清酸(5-foa)的cove-n培养基中。在5-foa的存在下,pyrg基因已经缺失的菌株将会生长;那些保留该基因的菌株会将5-foa转化为毒性中间体5-氟-ump。分离出生长更加迅速的菌落。分离菌株命名为nn059207。构建用于在na1和酸稳定性淀粉酶基因座处引入frt位点和黑曲霉fcy1的phuda1067设计在两个位点处均引入xbai位点的以下引物bac-f和bac-r,以分离融合有frt-f和frt-f3位点的pbluescriptiisk-的载体序列:bac-f(seqidno:75):tctagagaataggaactcggaataggaacttcaagatgaattcgcggccgcgbac-r(seqidno:76):tctagattgaagttcctattccgagttcctattcttcaaatagtataggaacttcagcatgcaagcttggcctccgc以pbluescriptiisk-作为模板,使用引物对bac-f和bac-r进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除2.7kb的产物条带。将2.7kb的扩增dna片段用xbai消化,并连接到用xbai消化的phuda1078,以创建phuda1078-na2。设计在两个位点处均引入paci位点的以下引物flp-f和flp-r,分离由构巢曲霉木聚糖酶启动子(pxlna)和米曲霉niad终止子(tniad)驱动的flp表达盒:flp-f(seqidno:77):ttaattaatggaagtgcgttgatcattattflp-r(seqidno:78):ttaattaaactagtggagcgaaccaagtga以phuda996作为模板,使用引物对flp-f和flp-r,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除2.4kb的产物条带。将2.4kb的扩增dna片段用paci消化,并连接到用paci消化的phuda1078-na2中,以创建phuda1067。质粒图谱显示于图9。在黑曲霉nn059207的na1和酸稳定性淀粉酶基因座处引入frt位点和黑曲霉fcy1基因和t.c.ga基因将phuda1067引入到黑曲霉菌株nn059207中。从补充有1%d-木糖的cove-n(tf)中选择转化子。将随机选择的转化子接种到cove-n平板上。将在cove-n平板上生长良好的菌株进行纯化,并进行southern印迹分析,以确认phuda1067中的frt位点和fcy1基因是否在na1和酸稳定性淀粉酶基因座处正确引入。使用用于非放射性探针的以下引物组来分析所选择的转化子。对于t.c.ga编码区:正向引物(seqidno:79):tcgagtgcggccgacgcgtacgtc反向引物(seqidno:80):cagagagtgttggtcacgta将从所选择的转化子中提取的基因组dna用hindiii消化。通过正确的基因引入事件,当如上所述用探针探测时,观察到hindiii消化的8.7kb(na1)、7.2kb(酸稳定性淀粉酶)和5.6kb(na2)大小的杂交信号。在具有正确的3个拷贝整合事件的菌株中,选择称作nn059208的菌株。图10示出nn059208中的示意性na1基因座(上)、na2基因座(中)和酸稳定性淀粉酶基因座(下)。将分别具有1个拷贝和3个拷贝t.c.ga基因的nn059203和nn059208在摇瓶中发酵,并按照上述材料和方法测量其酶活(agu活性);结果示于以下表1。通过用phuda1000转化nn059183或c1997而生成的双拷贝t.c.ga菌株(1000-7、1000-18)也进行发酵。菌株宿主质粒t.c.ga拷贝agu相对活性nn059203nn059183phuda107811.001000-7nn059183phuda100021.98~2.081000-18c1997phuda100021.96~2.10nn059208nn059203phuda106732.87~3.00表1.1个、2个和3个拷贝菌株的agu活性,其中将nn059203标准化为1.00。实施例9.通过flp在nn059208的3个基因座(na1、na2和酸稳定性淀粉酶)中将t.c.ga基因同时基因交换为ja126淀粉酶基因nn059208中pyrg基因拯救首先,如下对在nn059208的na1和酸稳定性淀粉酶基因座处引入的pyrg基因进行拯救。将菌株nn059208接种一次到含有10mm尿苷和1g/l5-氟-乳清酸(5-foa)的cove-n培养基中。在5-foa的存在下,pyrg基因已经缺失的菌株将会生长;那些保留该基因的菌株会将5-foa转化为毒性中间体5-氟-ump。分离出生长更加迅速的菌落。分离的菌株命名为nn059209。构建用于在三个基因座处引入ja126淀粉酶基因的prika147通过nhei消化,从phuda1067中移除含有黑曲霉fcy1基因的1.5kbdna片段。将回收的1.5kb片段重连。将连接混合物转化到大肠杆菌dh5α中,以创建表达质粒phuda1067-fcy。设计分别引入bamhi位点和pmli位点的以下引物126-f和126-r,来分离ja126淀粉酶的编码区,该编码区包含黑曲霉酸稳定性淀粉酶的分泌信号序列、微小根毛霉(rhizomucorpusillus)淀粉酶的催化结构域和黑曲霉葡糖淀粉酶的接头和淀粉结合域:126-f(seqidno:81):ggatccaccatgcggctctccacatcc126-r(seqidno:82):cacgtgtgattacggacacaatccgttattja126淀粉酶基因的核苷酸序列显示于seqidno:83,且所编码的氨基酸序列显示于seqidno:84。以pja126an作为模板,使用引物对126-f和126-r进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并从凝胶上切除1.9kb的产物条带。将1.9kb的扩增dna片段用bamhi和pmli消化,并连接到用bamhi和pmli消化的phuda1067-fcy,以创建prika147。质粒图谱示于图11。在nn059209中三个基因座(na1、na2和酸稳定性淀粉酶)中同时引入ja126淀粉酶基因将prika147引入到黑曲霉菌株nn059209中。从补充有1%d-木糖和10μg/ml5-氟胞嘧啶(5fc)的cove-n(tf)中选择转化子。将随机选择的转化子接种到补充有10μg/ml5-氟胞嘧啶(5fc)的cove-n平板上。将在补充有10μg/ml5-氟胞嘧啶(5fc)的cove-n平板上生长良好的菌株进行纯化,并进行southern印迹分析,以确认prika147中的ja126基因是否正确引入到na1、na2和酸稳定性淀粉酶基因座处。使用用于非放射性探针的以下引物组来分析所选择的转化子。对于ja126编码区:正向引物(seqidno:85):tcgaacttcggcgacgagtcgcagttgaa反向引物(seqidno:86):cccaacatctcggaaatcctggagaaaccc将从所选择的转化子中提取的基因组dna用hindiii和pmli消化。通过正确的基因引入事件,当用上述探针探测时,观察到hindiii和pmli消化的8.0kb(na1)、6.5kb(酸稳定性淀粉酶)和4.8kb(na2)大小的杂交信号。图12示出在prika147正确整合到nn059208中之后的示意性na1(上)、na2(中)和酸稳定性淀粉酶基因座(下)。通过补充有10μg/ml5-氟胞嘧啶(5fc)的cove-n平板的转化子产生频率是通过不含5fc的cove-n平板的频率的约1/10,000。但是,通过补充有10μg/ml5-氟胞嘧啶(5fc)的cove-n平板产生的菌株的50%给出在三个基因座的正确整合,而通过不含5fc的cove-n平板随机选择的所有菌株最多给出在一个基因座处的正确整合,而没有5fc时所产生的菌株均不显示出正确的整合事件。这表明使用fcy1基因的逆选择效果非常好。将在三个基因座处引入ja126淀粉酶的三个菌株(r147-17、r147-26、r147-34)在摇瓶中发酵,并按照上述的材料和方法测量其酶活(afau活性);结果示于以下表2。作为参照,对通过常规同源重组产生的单拷贝ja126淀粉酶菌株c2325(未示出)也加以发酵。菌株ja126拷贝afau相对活性c232511.00r147-1732.75~2.96r147-2632.82~3.00r147-3433.15~3.18表2.单拷贝菌株和3拷贝菌株的afau活性,其中c2325标准化为1.00。实施例10.在米曲霉jal1338的淀粉酶b(amyb)基因座处引入frt位点和tk基因构建ligd破坏质粒pjal1123分别破坏pdv8中bamhi和bglii的两个限制识别位点。首先将pdv8用bamhi消化,然后通过用klenow酶和四种dntp处理来将末端完全补平。将得到的6030bp片段再连接,得到质粒pjal504。第二步,将pjal504用bglii消化,然后通过用klenow酶和四种dntp处理将末端完全补平。将得到的6034bp片段再连接,得到质粒pjal504-δ-bglii。通过使用引物172450和172449进行的pcr,扩增出含有两侧为构巢曲霉gpd启动子和trpc终止子的hsv-tk基因的2522bp片段。然后将pcr片段克隆到质粒4blunt载体中,得到pjal574。引物172449(seqidno:87):gacgaattccgatgaatgtgtgtcctg引物172450(seqidno:88):gacgaattctctagaagatctctcgaggagctcaagcttctgtacagtgaccggtgactc将来自pjal554的米曲霉pyrg基因分离成2403bpstui-ecori片段,其中ecori位点通过用klenow酶和4种dntp处理而完全补平。将片段克隆到pjal574中的唯一pmei位点中,得到质粒pjal1022。将质粒pjal1022用sspb1消化,并将8574bp片段分离并再连接,得到质粒pjal1025。将质粒pjal1025用ecori消化,将8559bp片段分离并再连接,得到质粒pjal1027。通过用bamhi部分消化,然后用klenow酶和四种dntp处理从而将末端完全补平,破坏两个bamhi位点中的一个位点。将8563bp片段再连接,得到质粒pjal1029。根据公开可得的米曲霉rib40基因组序列(nite数据库,http://www.bio.nite.go.jp/dogan/project/view/ao),设计出引物,以pcr扩增ligd基因(ao090120000322)的5’侧翼和3’侧翼序列。将用于5’侧翼部分的引物x4407c0和x4407c07分别用bamhi和ecori位点作为尾部:引物x4407c0(seqidno:89):cagggatccgtctaggctgcaataggc引物x4407c07(seqidno:90):ggagaattcggtcacatc用于3’侧翼部分的引物x7164d09和x7164d10分别以hindiii和spei位点结尾:引物x7164d09(seqidno:91):gacactagtcgtcggcagcaccggtg引物x7164d10(seqidno:92):cagaagcttcagagtgaaatagacgcgg使用来自toc1512的基因组dna作为模板,进行pcr反应。将1114bp和914bp的所扩增5’和3’片段用bamhi-ecori和hindiii-spei消化,分别得到1102bp片段和902bp片段。将3’侧翼片段克隆到pjal1029的对应部位中,产生pjal1120。然后将5’侧翼片段克隆到pjal1120的对应部位中,得到pjal1123。构建ligd(-)米曲霉菌株jal1194如wo0168864中所述,将质粒pjal1123用spei线性化,并用于转化米曲霉toc1512,在补充有0.6mm5-氟-2’-脱氧尿苷(fdu)的基本培养基上选择转化子。将多个转化子再分离两次,制备基因组dna。将来自各个转化子的染色体dna用asp718消化,并使用来自pjal1123的含有米曲霉ligd基因5’侧翼的1102bp32p-标记的dnaecori-bamhi片段作为探针,通过dna印迹加以分析。通过3828bpasp718条带的消失和2899bpasp718条带的出现来识别目标菌株。将具有上述特征的一个转化子命名为jal1194。分离pyrg(-)米曲霉菌株jal1196将米曲霉菌株jal1194筛选对5-氟-乳清酸(foa)的抗性,以在补充有1.0m蔗糖作为碳源、10mm硝酸钠作为氮源以及0.5mg/mlfoa的基本平板(coved.j.1966.biochem.biophys.acta.113:51-56)上识别自发的pyrg突变体。一菌株jal1196被鉴定为pyrg(-)。jal1196是尿苷依赖型,因此可以对其转化野生型pyrg基因,并通过在缺乏尿苷的情况下的生长能力来选择转化子。构建aflatrem基因簇(atm)缺失质粒pjal1202通过用引物t5483h12和t5483g10对pjal574进行pcr,将米曲霉端粒序列引入到tk表达盒周围:引物t5483h12(seqidno:93):gcacatatgatttaaatccctaatgttgaccctaatgttgaccctaatgttgagcggccgcgtttaaacgaattcgccc引物t5483g10(seqidno:94):cgtaagcttatttaaatccctaatgttgaccctaatgttgaccctaatgttgagaccggtgactctttctg将扩增的2595bp片段用ndei和hindiii消化,将得到的2582bp片段克隆到pu19中的对应部位处,产生pjal835。将质粒pjal835用hindiii消化,通过用klenow酶和四种dntp处理将末端补平,然后再连接,生成pjal955。将质粒pjal554用hindiii和asp718消化,将得到的编码米曲霉pyrg基因的1994bp片段克隆到ptoc65中的对应部位处,产生pjal1183。通过引物d5831f08和d5831f09从toc1512基因组dna中扩增出1535bp的atm5’片段:引物d5831f08(seqidno:95):gacgaattcggcgtgggaaattcctgg引物d5831f09(seqidno:96):ccctacacctggggtacc将扩增的片段用ecori和asp718消化,将得到的1514bp片段克隆到pjal1183中对应部位处,产生pjal1194。将来自pjal1194的含有atm5’侧翼和pyrg基因的3529bpecori-noti片段与来自pjal955的含有tk基因的3529bp片段连接在一起,产生pjal1202。质粒pjal1202是用于使染色体atm基因簇缺失的质粒。构建atm(-)米曲霉菌株jal1268将质粒pjal1202用spei线性化,并用于转化米曲霉jal1196。如wo0168864中所述,在补充有0.6mm5-氟-2’-脱氧尿苷(fdu)的基本培养基上选择转化子。将多个转化子再分离两次,并制备基因组dna。将来自各个转化子的染色体dna用saci消化,并使用来自pjal1194的含有米曲霉atm基因簇5’侧翼的1514bp32p标记的dnaecori-asp718片段作为探针,通过southern印迹加以分析。通过3230bpsaci条带的消失和4436bpsaci条带的出现,识别出目标菌株。将具有上述特征的一个转化子命名为jal1268。分离pyrg(-)米曲霉菌株jal1338将米曲霉菌株jal1268筛选对5-氟-乳清酸(foa)的抗性,以在补充有1.0m蔗糖作为碳源、10mm硝酸钠作为氮源以及0.5mg/mlfoa的基本平板(coved.j.1966.biochem.biophys.acta.113:51-56)上识别自发的pyrg突变体。一个菌株jal1338被识别为pyrg(-)。jal1338是尿苷依赖型的,因此可以对其转化野生型pyrg基因,并通过在缺少尿苷的情况下的生长能力来选择转化子。构建用于在淀粉酶b基因座处整合的含有侧翼为frt位点的tk基因的质粒pjal1258根据公开可得的米曲霉rib40基因组序列(nite数据库,http://www.bio.nite.go.jp/dogan/project/view/ao),设计出引物,以扩增淀粉酶b(amyb)基因(ao090023000944)的5’侧翼和3’侧翼序列。用于5’侧翼部分的引物d5775f04和d5775d07分别以noti和hindiii位点结尾:引物d5775f04(seqidno:97):gacgcggccgcgctttgctaaaactttgg引物d5775d07(seqidno:98):gacaagcttatgctcgatggaaacgtgcac用于3’侧翼部分的引物d5775d08和d5775f05分别以hindiii和noti位点结尾:引物d5775d08(seqidno:99):gacaagcttacagtagttggactactttac引物d5775f05(seqidno:100):gacgcggccgcgacgagcaactgacggc使用来自toc1512的基因组dna作为模板进行pcr反应。将1307bp和511bp的所扩增5’和3’片段用noti和hindiii消化,分别得到1294bp片段和498bp片段。然后将5’和3’侧翼片段克隆到ptoc65的noti位点,得到pjal1196。通过引物f3-1和f3-2的退火而形成具有用于克隆到puc19的限制性酶切位点bamhi和psti中的悬突的接头,将酵母2μ质粒frt位点f和f3(schlaket.andbodej.useofmutatedflprecognitiontarget(frt)sitesfortheexchangeofexpressioncassettesatdefinedchromosomalloci.biochemistry33:12746-12751)克隆到puc19中,得到pjal952:引物f3-1(seqidno:101):gatccttgaagttcctattccgagttcctattcttcaaatagtataggaacttcactgca引物f3-2(seqidno:102):tgaagttcctatactatttgaagaataggaactcggaataggaacttcaa通过测序来证实frtf3位点插入到puc19中。然后将引物f-1和f-2一起退火,形成具有用于克隆到pjal952的限制性酶切位点asp718的悬突的接头:引物f-1(seqidno:103):gtaccttgaagttcctattccgagttcctattctctagaaagtataggaacttca引物f-2(seqidno:104):gtactgaagttcctatactttctagagaataggaagtcggaataggaacttcaa通过测序来证实frtf位点以与f3相同的方向插入pjal952中,正确的克隆命名为pjal953。通过从含有frt位点f和f3的pjal963中获取142bpsaci-hindiii片段,并将其克隆到用saci-hindiii消化的pjal1196中,来将frtf-f3位点插入到amyb侧翼之间,得到含有5’amyb侧翼并接有frtf-f3位点和3’amyb侧翼的pjal1249。然后,按照如下将pyrg和tk基因插入到frtf和frtf3位点之间。将末端通过用klenow酶和四种dntp处理而补平的pjal1029的4838bphindiii-sspbi片段克隆到pjal1249的smai位点中,得到具有以下不同元件的排列的质粒:5’amyb侧翼–frtf–pyrg–tk–ftrtf3–3’amyb侧翼,将其命名为pjal1258。构建frt、pyrg和tk整合在amyb基因座的米曲霉菌株jal1386将质粒pjal1258用noti线性化,并用于转化米曲霉jal1338;在基本培养基上选择转化子。将多个转化子再分离两次,制备出基因组dna。将来自各个转化子的染色体dna用xhoi消化,并使用来自pjal1196的含有米曲霉amyb基因5’侧翼的1294bp32p标记的dnanoti-hindiii片段作为探针,通过southern印迹加以分析。通过4164bpxhoi条带的消失和8971bpxhoi条带的出现,来识别目标菌株。将具有上述特征的一个转化子命名为jal1386。分离pyrg(-)米曲霉菌株jal1394将米曲霉菌株jal1386筛选对5-氟-乳清酸(foa)的抗性,以在补充有1.0m蔗糖作为碳源、10mm硝酸钠作为氮源以及0.5mg/mlfoa的基本平板(coved.j.1966.biochem.biophys.acta.113:51-56)上识别自发的pyrg突变体。一菌株jal1394被识别为pyrg(-)。jal1394是尿苷依赖型的,因此可以用野生型pyrg基因对其进行转化,并通过在缺乏尿苷的情况下的生长能力来选择转化子。实施例11.通过flp向jal1394的amyb基因座进行位点特异性整合构建埃默森篮状菌(talaromyceemersonii)amg表达盒prika99对含有内含子的埃默森篮状菌amg基因进行优化,提供用于在曲霉中表达的合成基因(seqidno:105)。出于克隆的目的,分别向5’端和3’端添加bamhi和xhoi限制性酶切位点。基于质粒pj241:13509-huda2的序列,获得合成的基因。分别从质粒pj241:13509-huda2和phuda1000中分离编码埃默森篮状菌amg基因的2085bpbamhi-xhoi片段和9510bpbamhi-xhoi片段。将两个片段连接在一起,以创建prika99。通过flp将prika99位点特异性整合在jal1394中将prika99引入到米曲霉菌株jal1394中。在补充有1%d-木糖和0.6mm5-氟-2’-脱氧尿苷(fdu)的kcl-平板上选择转化子。将四个转化子再分离两次,制备出基因组dna。将来自各个四种转化子的染色体dna用bglii-draiii和bglii-kspi消化,并首先使用来自prika99的含有amg基因的2095bp32p标记的dnabamhi-xhoi片段作为探针,其次在去除滤器之后通过使用来自prika99的含有构巢曲霉xlna启动子的731bp32p标记的dnaafei-paci片段作为探针,通过southern印迹进行分析。正确的整合事件通过以下来识别:1)使用amg探针:在bglii-draiii消化中的7145bp和3739bp条带,以及bglii-kspi消化中的6845bp条带;2)使用构巢曲霉xlna启动子探针:bglii-draiii消化中的6845bp条带,以及bglii-kspi消化中的4039bp条带。实施例12.经由5-氟胞嘧啶(5fc)的米曲霉生长抑制以及胞嘧啶氨基酶的破坏为测试米曲霉生长受5-氟胞嘧啶(5fc)的抑制,将bech2的孢子划线接种在补充有不同浓度5fc(2.5、1.5和0.625μg/ml)的cove-n(tf)上。在最低的5fc浓度(0.625μg/ml)下,检测不到生长,表明米曲霉也具有胞嘧啶脱氨酶。在米曲霉中,仅有一种与黑曲霉fcy1基因(embl:am269962)直系同源的基因(公共基因组序列的ao090003000802),因此将其破坏以证实该基因是导致细胞在5fc上生长时死亡的胞嘧啶脱氨酶。通过使用在nielsen等人(2005)efficientpcr-basedgenetargetingwitharecyclablemarkerforaspergillusnidulans,fungalgentbiol43:54-64中记载的双分基因(bipartitegene)靶向底物来破坏ao090003000802。通过pcr扩增生成含有米曲霉ao090003000802基因5’侧翼和部分pyrg基因(pyrg基因的启动子和2/3编码区)的2145bp片段。首先,以bech2基因组dna为模板,使用引物ojal132:cagatactggttccttacgg(seqidno:108)和ojal133:cgtccacgcggggattatgcgtagaatgcagagatagctg(seqidno:109),通过pcr扩增出含有ao090003000802的5’侧翼的1036bp片段。然后,以pjal554为模板dna,使用引物x1111c07:gcataatccccgcgtggacg(seqidno:110)和ojal114:ccaacagccgactcaggag(seqidno:111),通过pcr扩增含有pyrg5’部分的1129bp片段。将所扩增的产物在1.0%琼脂糖凝胶上分离,并混合在一起,使用引物ojal132和ojal114进行pcr,得到2145bp的扩增产物,将其在1.0%琼脂糖凝胶上纯化。通过pcr扩增,生成含有米曲霉ao090003000802基因3’侧翼和部分pyrg基因(pyrg基因的2/3编码区和终止子)的2436bp片段。首先,以bech2基因组dna为模板,使用引物ojal134:cgataagctccttgacggggttgagcactgcttttggatc(seqidno:112)和ojal135:gctcacccggcataagttgc(seqidno:113),通过pcr扩增含有ao090003000802的5’侧翼的1011bp片段。然后,以pjal554为模板dna,使用引物x1111c08:ccccgtcaaggagcttatcg(seqidno:114)和ojal113:gagctgctggatttggctg(seqidno:115),通过pcr扩增含有pyrg5’部分的1445bp片段。将扩增的产物在1.0%琼脂糖凝胶上分离,并混合在一起,使用引物ojal1135和ojal135进行pcr,生成2436bp的扩增产物,将其在1.0%琼脂糖凝胶上纯化。为破坏ao090003000802基因,将上述两种扩增的2145bp和2436bp片段进行混合,并转化到米曲霉jal1398菌株中,从cove-n平板上选择转化子。使用southern印迹分析来证实ao090003000802基因的破坏。将提取自20个转化子的基因组dna用pvui-spei消化,并使用32p标记的上述扩增pcr1036bp片段作为探针,进行southern印迹分析。通过5.5kbpvui-spei条带的消失和6.9kbpvui-spei条带的出现,来识别目标菌株。同时,测试菌株在含有0.625μg/ml5fc的cove-n平板上的生长情况,仅有在6.9kb具有预计条带的菌株显示出生长,这表明ao090003000802基因是胞嘧啶脱氨酶.。在这些菌株中,选择一种并命名为jal1500。实施例13.在黑曲霉nn059209中pay(推定的烷基型硫酸酯酶)基因座处引入frt位点和黑曲霉fcy1基因构建用于在pay基因座处引入frt位点和黑曲霉fcy1的phuda1174(图13)基于embl:am270278中的核苷酸序列信息,设计分别引入xbai位点和pmei位点的下列引物3pay-f和3pay-r,以分离黑曲霉pay基因的3'侧翼区:3pay-f:ttgcttctagacttctatttcctaatat(seqidno:116)3pay-r:ttgtttaaacttaattaaccgcgccat(seqidno:117)使用3pay-f和3pay-r引物对,以黑曲霉菌株nn059183的基因组dna作为模板,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并将2.1kb产物条带从凝胶上切离。2.1kb扩增的dna片段用xbai和pmei消化,并连接到用xbai和pmei消化的phuda801以产生phuda801-3pay。基于embl:am270278和dj052242中的核苷酸序列信息,设计分别地引入noti位点和spei位点的下列引物5pay-f和5pay-r,以分离融合有frt-f位点的黑曲霉pay基因的5'侧翼区:5pay-f:ggtggcggccgcgccgacggtgctggagga(seqidno:118)5pay-r:tttactagtgaagttcctatactttctagagaataggaactcggaataggaacttcaagatgaattcctagtcgg(seqidno:119)使用5pay-f和5pay-r引物对,以黑曲霉菌株nn059183的基因组dna作为模板,进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,并将1.3kb产物条带从凝胶上切离。该1.3kb扩增的dna片段用noti和spei消化,并连接到用noti和spei消化的phuda801-3pay以产生phuda801-3pay-5pay。通过nhei和xbai消化,从phuda440-frt中回收包含t.c.ga基因的4.3kbdna片段,该t.c.ga基因由三重串联na2启动子(pna2)和amg终止子(tamg)驱使。将该回收的4.3kb片段连接到nhei和xbai消化的phuda801-3pay-5pay。将连接混合物转化入大肠杆菌dh5a以构建表达质粒phuda801-3pay-5pay-tc。基于embl:embl:m19132和dj052242中的核苷酸序列信息,设计分别地引入xbai位点和spei位点的下列引物pyrg-f和pyrg-r,以分离融合有frt-f3位点的构巢曲霉pyrg基因。pyrg-f:ttagtactttgaagttcctattccgagttcctattcttcaaatagtataggaacttcaactagctagtgcatgcctagtggagcg(seqidno:120)pyrg-r:aagtctagaagcaagggcgaattccagca(seqidno:121)使用pyrg-f和pyrg-r引物对,以phuda794的基因组dna作为模板进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,且将2.1kb产物条带从凝胶上切离。该2.1kb扩增的dna片段用xbai和spei消化,并连接到用xbai消化的phuda801-3pay-5pay-tc以创造phuda801-3pay-5pay-tc-pyrg。基于embl:am269962中的核苷酸序列信息,设计在两个位点都引入nhei位点的下列引物fcy-f和fcy-r,以分离黑曲霉fcy1基因的整个区域。fcy-f:gctagcgcgaggctatcacggaggctgtgg(seqidno:122)fcy-r:gctagcttctgtggttcttgccatgatcgt(seqidno:123)使用fcy-f和fcy-r引物对,以黑曲霉菌株nn059183的基因组dna作为模板进行pcr反应。将反应产物在1.0%琼脂糖凝胶上分离,且将1.5kb产物条带从凝胶上切离。该1.5kb扩增的dna片段用nhei消化,并连接到用nhei消化的phuda801-3pay-5pay-tc-pyrg以产生phuda1174(图13)。在黑曲霉nn059209的pay基因座处引入frt位点和黑曲霉fcy1基因将phuda1174引入到黑曲霉菌株nn059209中。从cove-n(tf)选取转化子。将随机选取的转化子接种至带有2.5μm5-氟-2-脱氧尿苷(fdu)的cove-n平板上。将在具有2.5μμfdu的cove-n平板上生长良好且在具有10μg/ml5-氟胞嘧啶(5fc)的cove-n平板上几乎不生长的菌株纯化,并对其采取southern印迹分析以确定frt位点和fcy1基因是否正确地引入到pay位点。使用用于制作非放射性探针的下列引物组来分析所选的转化子。对于t.c.ga编码区域,正向引物:tcgagtgcggccgacgcgtacgtc(seqidno:124),反向引物:cagagagtgttggtcacgta(seqidno:125)。从所选转化子中提取的基因组dna用pmli消化。通过正确的基因引入事件,如上所述,观察探测到经pmli消化的7.7kb大小的杂交信号。在给予正确的整合事件的这些菌株之中,选取菌株nn059280。实施例14.用于产生具有改变的ja126淀粉酶基因拷贝的菌株的竞争性基因交换在nn059280中拯救pyrg基因对在nn059280中pay基因座处引入的pyrg基因作如下拯救。将该菌株nn059280一次接种至含有10mm尿苷和1g/l5-氟-乳清酸(5-foa)的cove-n培养基。其中已缺失pyrg基因的菌株将在5-foa的存在下生长;保留该基因的那些菌株会将5-foa转化为有毒的中间体5-氟-ump。分离生长更快的菌落。将分离的菌株命名为m1146。空载体phuda1306(图15)的构建用nhei和pmli消化prika147。通过t4dna聚合酶补平并重连8.1kbdna片段。将生成的质粒称为phuda1306(图15)。用prika147和phuda1306进行竞争性基因交换以生成具有改变的ja126淀粉酶基因拷贝的菌株;参见图16a-16c。将prika147和phuda1306共同引入到黑曲霉菌株m1146。从补充有1%d-木糖和10μg/ml5-氟胞嘧啶(5fc)的cove-n(tf)上选取转化子。将随机选取的转化子接种至补充有10μg/ml5-氟胞嘧啶(5fc)的cove-n平板。纯化在补充有10μg/ml5-氟胞嘧啶(5fc)的cove-n平板上生长良好的菌株并对其采取southern印迹分析,以确定prika147中的ja126基因是否正确地引入到na1、na2、sp288或pay基因座。用制作非放射性探针的下列引物组来分析所选转化子。对于ja126编码区:正向引物:tcgaacttcggcgacgagtcgcagttgaa(seqidno:126)反向引物:cccaacatctcggaaatcctggagaaaccc(seqidno:127)从所选转化子中提取的基因组dna用hindiii和pmli消化。通过正确的基因引入事件,如上所述,观察探测到经hindiii和pmli消化的8.0kb(na1)、6.5kb(sp288)、4.8kb(na2)和4.5kb(pay)大小的杂交信号。产生具有0~4个ja126基因拷贝的转化子的频率大部分是相同的。因此,通过表达质粒和空质粒的共引入,很容易获得具有不同目标基因拷贝数的转化子。实施例15.通过flp及其热稳定变体flpe将nn059280中4个基因座(na1、na2、sp288和pay)处的t.c.ga基因同时基因交换成ja126淀粉酶基因热稳定flp变体(flpe)表达载体phuda1352的构建基于文献中flpe的序列信息(improvedpropertiesofflprecombinaseevolvedbycyclingmutagenesisf.buchholz,p.o.angrand,a.f.stewart.nat.biotechnol.,16(1998),657-662页),制备如下引物。flpe1:ggatctaccatgtcccagttcgatatcctctgcaagaccccccccaaggtcctcgtccgccagttcgtcgagcgcttcgagcgcccctccggcgagaagatcgcctcctgcgccg(seqidno:128)flpe2:atgcttctggccgttgtaggggatgatggt(seqidno:129)flpe3:accatcatcccctacaacggccagaagcat(seqidno:130)flpe4:ttgatggcgaagatggggtagggggcgttc(seqidno:131)flpe5:gaacgccccctaccccatcttcgccatcaa(seqidno:132)flpe6:ttcggatcagatgcggcggttgatgtagga(seqidno:133)使用引物对flpe1和flpe2,flpe3和flpe4及flpe5和flpe6,以phuda996作为模板进行pcr反应。在1.0%琼脂糖凝胶上分离反应产物,并将0.3、0.6和0.5kb产物带从凝胶上切离。混合这三个片段并用于运用引物对flpe1和flpe6进行的第二轮pcr反应。在1.0%琼脂糖凝胶上分离反应产物,并将1.3kb产物带从凝胶上切离。1.3kb扩增的dna片段用bamhi和bstbi消化,并连接到用bamhi和bstbi消化的phuda996以产生phuda1352。构建携带热稳定flp变体(flpe)的ja126淀粉酶表达质粒,表达载体phuda1356。用bamhi和bstbi消化phuda1352。将1.3kbdna片段连接到用bamhi和bstbi消化的prika147以生成phuda1356(图17)。flp和flpe之间同时基因交换效率的比较将prika147和phuda1356引入到黑曲霉菌株m1146。从补充1%d-木糖和10μg/ml5-氟胞嘧啶(5fc)的cove-n(tf)上选取转化子。将随机选取的转化子接种至补充有10μg/ml5-氟胞嘧啶(5fc)的cove-n平板上。纯化在补充有10μg/ml5-氟胞嘧啶(5fc)的cove-n平板上生长良好的菌株,并对其进行southern印迹分析,以确认prika147中的ja126基因是否正确地引入到na1、na2、sp288或pay基因座。用制作非放射性探针的下列引物组来分析所选的转化子。对于ja126编码区:正向引物:tcgaacttcggcgacgagtcgcagttgaa(seqidno:134)反向引物:cccaacatctcggaaatcctggagaaaccc(seqidno:135)用hindiii和pmli消化从所选转化子中提取的基因组dna。通过正确的基因引入事件,如上所述,观察探测到经hindiii和pmli消化的8.0kb(na1)、6.5kb(sp288)、4.8kb(na2)和4.5kb(pay)大小的杂交信号。使用flpe(phuda1356)进行同时整合的频率比使用flp(prika147)时的频率高约3倍,因此热稳定flp变体flpe提供提高的基因座特异性整合频率。实施例16.在米曲霉中amyb和#13基因座处引入frt位点和tk基因构建米曲霉菌株jal1398niad-米曲霉菌株jal828的分离首先,对于米曲霉菌株5-58(wo20099106488)筛选氯酸盐抗性,从而在补充有1.0m蔗糖作为碳源、10mm谷氨酸钠作为氮源和5%氯酸盐的基本平板(minimalplate)上鉴定自发的niad突变体(coved.j.1966.biochem.biophys.acta.113:51-56)。一个菌株jal828被鉴定为niad-。第二,对于米曲霉菌株jal828筛选5-氟-乳清酸(foa)抗性,从而在补充有1.0m蔗糖作为碳源、10mm硝酸钠作为氮源和0.5mg/mlfoa的基本平板上鉴定自发pyrg突变体(coved.j.1966.biochem.biophys.acta.113:51-56)。一个菌株cols454被鉴定为pyrg-。cols454是尿苷依赖型,因此,可以对其转化野生型pyrg基因,并根据在尿苷不存在的情况下的生长能力选择转化子。第三,如在实施例10中所描述的,将米曲霉cols454菌株制成ligd-型,得到米曲霉菌株jal1390。第四,如前所述,将米曲霉菌株jal1390制成pyrg-型,得到米曲霉菌株jal1398。构建具有在基因座amyb和#13处整合的frt::tk的米曲霉菌株jal1523为了整合侧翼是frt位点的tk,用noti将质粒pjal1258线性化,并将其用于转化米曲霉jal1398;在基本培养基上选取转化子。将若干个转化子再分离两次,并准备基因组dna。用xhoi消化各个转化子的染色体dna,并使用pjal1196的包含米曲霉amyb基因的5'侧翼的1294bp32p标记的dnanoti-hindiii片段作为探针,通过southern印迹对其进行分析。用4164kbxhoi条带的消失和8971kbxhoi条带的出现来鉴定目标菌株。将一个具有以上特征的转化子命名为jal1450。分离pyrg-型米曲霉菌株jal1467对于米曲霉菌株jal1450筛选5-氟-乳清酸(foa)抗性,从而在补充有1.0m蔗糖作为碳源、10mm硝酸钠作为氮源和0.5mg/mlfoa的基本平板上鉴定自发的pyrg突变体(coved.j.1966.biochem.biophys.acta.113:51-56)。一个菌株jal1467被鉴定为pyrg-型。jal1467是尿苷依赖型,因此,可以对其转化野生型pyrg基因,并根据在尿苷不存在的情况下的生长能力选择转化子。构建包含侧翼是frt位点用于在#13基因座进行整合的tk基因的质粒pjal1313在质粒pjal835(us2010062491)中,通过用hindiii打开质粒,然后通过用4dntp's和klenow处理将端部补平,接着进行重连而破坏单个hindiii,生成质粒pjal955。在米曲霉rib40基因组序列之外(www.bio.nite.go.jp/dogan/project/view/ao),设计引物以扩增基因座#13的5'侧翼和3'侧翼序列。5'侧翼部分的引物,k6763e12:gacgcggccgccgcgtggaggtctaggac(seqidno:136)和k6763f01:gacaagcttacaaacccgtgacactcc(seqidno:137),分别以noti和hindiii位点结尾。3'侧翼部分的引物,k6763f02:gacaagcttacgcatgtatgtatgtgtc(seqidno:138)和k6763f03:gacgtttaaacggatgggtttgccatac(seqidno:139),分别以hindiii和pmei位点结尾。将toc1512的基因组dna用作pcr反应的模板。分别用noti-hindiii和hindiii-pmei消化1065bp和1032bp的扩增的5'和3'片段,分别得到1052bp片段和1021bp片段。然后将5'和3'侧翼片段克隆到pjal955中的noti-pmei位点中,得到pjal968。用nhei-pmei消化质粒pjal968,并通过dntp's和klenow的处理对端部完全补平。将4548bp片段纯化并自连,得到质粒pjal1285。通过引物f3-1(seqidno:15)和f3-2(seqidno:16)的第一退火处理,形成具有悬突的用于克隆到puc19的限制酶切位点bamhi和psti的接头,将酵母2μ质粒frt位点f和f3(schlaket.andbodej.useofmutatedflprecognitiontarget(frt)sitesfortheexchangeofexpressioncassettesatdefinedchromosomalloci.biochemistry33:12746-12751)克隆到puc19,得到pjal952。通过测序来检验frtf3位点插入到puc19中。第二,将引物f-1和f-2一起退火,以形成具有悬突的接头用于克隆到pjal952的限制酶切位点asp718。通过测序来检验frtf位点以与f3的方向相同的正确方向插入pjal952,并将正确的克隆命名为pjal953。用saci-scai消化质粒pjal953,并将生成的1866bp片段连接到pic19h的920bpsaci-scai片段,得到质粒pjal1289。为了在frt位点之间插入hsv-tk基因,将4839bphindiii-bsrgi克隆到用smai消化的pjal1289中,其中4839bphindiii-bsrgi的端部通过dntp's和klenow的处理被完全补平。按以下方式具有不同元件的质粒被命名为pjal1293:frtf_pyrg_hsv-tk_frtf3。将携带pjal1293的frtf_pyrg_hsv-tk_frtf3部分的4984bphindiii片段连接到来自pjal1285的4548bphindiii片段。按以下方式具有不同元件的质粒被命名为pjal1313:5'#13侧翼_frtf_pyrg_hsv-tk_frtf3_3'#13侧翼。构建具有在#13基因座处整合的frt、pyrg和tk的米曲霉菌株jal1523用noti将质粒pjal1313线性化,用于转化米曲霉jal1467,在基本培养基上选取转化子。将多个转化子再分离两次,制备基因组dna。将各个转化子的染色体dna用nhei-ndei消化,并使用pjal1313的包含米曲霉#13基因座的3'侧翼的893bp32p标记的dnancoi-hindiii片段作为探针,通过southern印迹对其进行分析。通过3896kbnhei-ndei条带的消失和5607kbnhei-ndei条带的出现来鉴定目标菌株。将一个具有以上特征的转化子命名为jal1523。分离pyrg-型米曲霉菌株jal1540对于米曲霉菌株jal1523筛选5-氟-乳清酸(foa)抗性,从而在补充有1.0m蔗糖作为碳源、10mm硝酸钠作为氮源和0.5mg/mlfoa的基本平板上鉴定自发的pyrg突变体(coved.j.1966.biochem.biophys.acta.113:51-56)。一个菌株jal1540被鉴定为pyrg-型。菌株jal1540是尿苷依赖型,因此,可以对其转化野生型pyrg基因,并根据在尿苷不存在的情况下的生长能力选择转化子。实施例17.利用frt/flp重组体系在里氏木霉中进行位点特异性整合培养基和试剂使用以下培养基和试剂:lb液体培养基+100μg/ml氨苄青霉素:每升10g胰蛋白胨,5g酵母提取物,5gnacl和1ml100mg/ml氨苄青霉素。2ytamp:每升16g胰蛋白胨,10g酵母提取物,5gnacl,15g细菌琼脂和1ml100mg/ml氨苄青霉素。cove:每升342.3g蔗糖,20mlcove盐溶液,10ml1m乙酰胺,10ml1.5mcscl及25g诺布尔琼脂。cove2+10mm尿苷:每升30g蔗糖,20mlcove盐溶液,10mm乙酰胺,15mmcscl及25g诺布尔琼脂。cim:每升20garbocel天然纤维素纤维(j.rettenmaierusalp),10g玉米浆固体(sigma),1.45g(nh4)2so4,2.08gkh2po4,0.28gcacl2,0.42gmgso4·7h20,0.42ml里氏木霉微量金属,2滴pluronicl61消泡剂,ph6.0。cove盐溶液:每升26gkcl,26gmgso4·7h2o,76gkh2po4,50mlcove微量元素。cove微量元素:每升0.004gna2b4o7·10h2o,0.4gcuso4·5h2o,1.2gfeso4·7h2o,0.7gmnso4·h2o,0.8gna2moo2·2h2o和10gznso4·7h2o。里氏木霉微量金属:216gfecl3·6h2o,58znso4·7h2o,27gmnso4·h2o,10gcuso4·5h2o,2.4gh3bo,336g柠檬酸。peg:每升500g聚乙二醇,10ml1mtrisph7.5,10ml1mcacl2。stc:每升0.5l1m山梨醇,10ml1mtrisph7.5,10ml1mcacl2。trmm:每升30g葡萄糖,0.6gcacl2,6g(nh4)2so4,20mlcove盐溶液,25g诺布尔琼脂。ypg2%:每升10g酵母提取物,20g蛋白胨,20g葡萄糖。质粒构建构建了frt位点整合载体pjfys147且展示于图18。构建了包含β-葡糖苷酶的frt/flp表达载体pjfys150且展示于图19。里氏木霉原生质体形成和转化如wo11/075677先前描述的那样产生里氏木霉菌株tv11的原生质体。在冰上解冻原生质体并将5×100μl原生质体小份转移到4×14mlfalcon2059管中。将pmei线性化的凝胶纯化的dna(约3μg)加至每个加有250μl60%peg的管中。轻轻地倒置5次来轻柔混合管中的内容物并在34℃温育30分钟。向每个管中加入3mlstc,向包含50mlpda+1m蔗糖的150mm平板中铺布1.5ml该内容物并用无菌的涂布器铺开。将平板在28℃温育约18小时,其后,加入20mlpda+10mm尿苷+35μg/ml潮霉素b(invitrogencat#10687010)的覆盖物。平板在28℃温育6天直到挑选转化子。挑选转化子用10μl接种环挑选转化子,转移到包含pda琼脂的75mm直径平板中,并在28℃温育5天。在cim培养基中进行转化子摇瓶分析用10μl接种环收集孢子并转移到125ml聚碳酸酯摇瓶,每个摇瓶含有25mlcim培养基并在28℃振荡下温育5天。修复hpt/tk标记(curehpt/tkmarkers)将7天平板培养物的孢子收集到0.01%吐温-20中,用血细胞计数器测定孢子浓度。在无菌dih2o中稀释孢子,以104、105和106铺布到150mmtrmm+2%葡萄糖平板+1μμ5-氟脱氧尿苷(fdu)。平板在28℃温育6天,用10μl接种环挑选孢子分离株,转移到新的pda平板并在28℃温育。基因组dna分离/southern分析将孢子收集到5ml0.01%吐温-20中,2ml用于接种至250ml带挡板摇瓶内的50mlypg2%培养基中。培养物在28℃170rpm振荡下温育40小时。去除琼脂块(agarplug),通过miraclothtm过滤培养物。用液氮冷冻所收获的生物质,并用研钵和研杵研磨菌丝。根据制造商的说明书使用plantmaxikit(qiagen,valencia,ca,usa)分离基因组dna,不同在于65℃的裂解温育期从10min延长到1.5小时。用nanodrop1000分光光度计(thermofischerscientific,waltham,ma,usa)测定所得的含dna溶液的浓度。在37℃用44单位的ndei在50μl反应体积中消化2.5μg基因组dna22小时。将消化产物在tae缓冲液中进行0.9%琼脂糖凝胶电泳。dna在凝胶中经0.25mhcl处理而片段化,用1.5mnacl-0.5mnaoh变性,用1.5mnacl-1mtris(ph8)中和,然后在20×ssc中用turboblottertm试剂盒转移到supercharge尼龙膜(两者都来自whatman,kent,uk)。使用uvstratalinkertm(stratagene,lajolla,ca,usa)将dnauv交联到膜上,并在42℃,在20mldigeasyhyb(rochediagnosticscorporation,indianapolis,in,usa)中预杂交1小时。用以下示出的正向和反向引物,根据制造商的说明书使用pcrdig探针合成试剂盒(rochediagnosticscorporation,indianapolis,in,usa)产生杂交到里氏木霉cbh2基因的3'侧翼的探针。pcr反应包括反应缓冲液(stratagene,lajolla,ca),400nm各引物,200μμdig标记的含dutp的dntp,125ngtv10基因组dna和1.5单位dna聚合酶。循环参数如下:1个循环:在95℃2分钟;25个循环:每个在95℃30秒,55℃30秒,和72℃40秒;和1个循环:在72℃7分钟。正向(069083):aaaaaacaaacatcccgttcataac(seqidno:140)反向(069084):aacaaggtttaccggtttcgaaaag(seqidno:141)将探针反应物在tae缓冲液中进行1%琼脂糖凝胶电泳,切除对应于探针的条带,并用凝胶提取试剂盒(qiageninc.,valencia,ca,usa)进行琼脂糖提取。将探针煮沸5分钟并加入到10mldigeasyhyb以产生杂交液。在42℃进行杂交15-17小时。然后在室温下高等严格条件下在2×ssc加0.1%sds中洗涤膜5分钟,接着在65℃下在0.1×ssc加0.1%sds中洗涤两次,每次15分钟。根据制造商的说明书使用化学发光检定(rochediagnostics,indianapolis,in,usa)检测探针-靶标杂交体。里氏木霉原生质体形成和flp载体的转化如先前描述的那样(14)产生里氏木霉菌株tv11的原生质体。在冰上解冻原生质体并将5×100μl原生质体各自转移到4×14mlfalcon2059管中。将pmei线性化的凝胶纯化的dna(大约2μg)加入到每个管中并加入250μl60%peg。轻轻地将管倒置5次来轻柔混合管中的内容物并在34℃温育30分钟。向每个管中加入3mlstc,向包含50mlpda+1m蔗糖的150mm平板中铺布1.5ml内容物并用一个无菌涂布器涂布开。将平板在28℃温育约18小时,其后加入20mlpda+10mm尿苷+35μg/ml潮霉素b(invitrogencat#10687010)的覆盖物。平板在28℃温育6天直到挑选转化子。孢子pcr为了筛选用于flp/frt载体整合的转化子,通过孢子pcr来筛选转化子。这通过用无菌的1μl接种环来收集孢子并将它们转移到0.6mleppendorf管中的25μlte缓冲液中来完成。用高功率微波处理孢子1分钟并立即将1μl加至advantagegcgenomiclapolymerasepcrmix中,advantagegcgenomiclapolymerasepcrmix包含以下组分:1×反应缓冲液,200μμdntp,400nm各引物,1.25u聚合酶。使用以下循环参数和下文所述的正向引物和反向引物扩增pcr产物用于5'整合或3'整合:95℃-10分钟,30个95℃-30秒,56℃-30秒,72℃-1分钟40秒的循环,和最后的72℃-7分钟的循环。5'重组正向(#0611526):ttcccttcctctagtgttgaat(seqidno:142)反向无整合(#0611527):tcgtcgaatactaacatcttgc(seqidno:143)反向整合(#0611528):cacggacctcgaacctttatat(seqidno:144)3'重组正向(#999661):cagcgagagcctgacctattgcatc(seqidno:145)反向无整合(#069084):aacaaggtttaccggtttcgaaaag(seqidno:146)反向整合(#0611648):gtggctgccgaggtgtgtatacca(seqidno:147)将整个pcr反应体系在1%琼脂糖胶上在包含500ng/ml溴化乙锭的50mltae缓冲液中跑胶并用uv光使产物可视化。结果设计frt位点整合载体,pjfys147(图18),从而使位点能cbh1基因座处整合到基因组中。两个frt位点略微不同,试图防止它们之间发生不希望的重组,它们分别被命名为用于5'和3'位点的frt-f和frt-f3。用于将载体靶向基因座的5'cbh1侧翼被选择为,使得cbh1编码序列以及启动子的1kb部分也会被缺失,因为启动子也被引入到随后使用的表达载体中并且成功的整合会修复启动子。由于载体在正确整合到cbh1基因座时还缺失了cbh1基因,这样可以进行简单的蛋白质组学筛选,因为sds-page图谱会随着cbh1的去除而实质性改变。当用pjfys147转化菌株tv11的原生质体时,得到133个转化子。挑取所有这些转化子并在纤维素酶诱导条件下进行摇瓶分析。在所分析的133个转化子中,两个表现出与cbh1缺失一致的蛋白组图谱。预期显示出改变的蛋白质组学图谱的两个转化子在cbh1基因座具有frt整合质粒。两个菌株jfys147-20和jfys147-73被铺布到含5-氟脱氧尿苷(fdu)的木霉属基本培养基平板,以试图促进hpt/tk盒的切除。在平板上从菌株jfys147-20得到72个抗fdu菌落并从菌株jfys147-73得到一个菌苔。从菌株jfys147-20挑取8个菌落,命名为jfys147-20a~jfys147-20h,并从jfys147-73挑取菌苔的一部分,所得的菌株命名为jfys147-73a。通过southern对4个jfys147-20的分离株和jfys147-73的一个分离区域进行分析,以确定frt盒是否已经明确整合且hpt/tk标记已被正确切除。southern分析显示,一个转化子即jfys47-20如预期的一样在cbh1基因座处具有删除盒,且生成的孢子后代已经切除了hpt/tk标记。另一个转化子jfys47-73未显示任何杂交。从jfys147-20b的基因组dna通过pcr扩增包含frtf和frtf3位点的区域,并进行测序以确认两个位点的存在。生成该菌株的原生质体,并用flp/frt整合载体pjfys150(图19)进行转化。表达载体pjfys150是带有用于潮霉素抗性的潮霉素磷酸转移酶基因的包含cbh1启动子和终止子的木霉属表达载体的衍生物。pjfys150与其母体不同的是,它还包含存在于jfys147-20b的基因组中的frt-f和frtf3位点以及内含子优化的衍生自prika147(从huda获得)的翻转酶基因(flp)盒。所运用的报告子是烟曲霉bg。用pmei将载体线性化以去除质粒的细菌繁殖部分,所得的凝胶纯化的片段用于转化jfys147-20b原生质体,得到20个转化子。所得的20个转化子在此代表2.5/μg的效率。通过扩增插入位点的5'区域,通过孢子pcr来分析20个转化子,以确定盒是否已经整合到期望的基因座。如果盒的整合是异位的,则产生1kb片段,如果整合发生在frt位点,则结果为1.8kbpcr产物。在得到的20个转化子中,18个似乎具有异位整合,而2个显示出与在cbh1基因座的整合一致的pcr条带,但是条带大小小于预期。当对pcr片段进行测序时,结果表明重组已经在基因座处的frtf位点之间和cbh1启动子内的两个不同区域发生。在转化之前,向原生质体储液中加入1%木糖,试图使需要的细胞反应事件加速。用与之前一样的表达载体pjfys150来转化带有额外木糖的原生质体,得到19个转化子。与之前一样,通过对5'端进行pcr筛选,还通过杂交至3'整合区域的额外引物组,来分析19个转化子。结果表明,19个转化子中有5个在5'区内的frtf位点处整合入盒。而且,2个转化子在pcr筛选中没有给出条带,意味着基因座的区域已经在转化期间重排,显示出一定程度的不精确的基因座特异性靶向,如在先前的转化子集合中看到的那样。对转化整合载体3'端进行frtf3位点区域pcr筛选,表明5个转化子已经经历了在3'端的flp介导的必要整合,且5个转化子中有3个已经经历了两个必要的重组。当单独分析每个区域时,一些转化子已经仅一个frt位点经历了期望的重组,但在另一个位点经历了非特异性重组。3个转化子在两个位点经历了所需的重组,这与没有添加木糖到原生质体存储培养基的过程相比具有改进。因此,在里氏木霉中成功地运用了flp/frt体系,以向里氏木霉的cbh1基因座引入表达质粒。具体地,生成菌株jfys147-20b,并且在cbh1基因座处包含frt位点。还产生了新的表达载体,其并入有cbh1启动子和终止子、作为选择标记的hpt基因、作为报告子的烟曲霉bg基因及体系所需的frt位点和flp基因。这个新载体,pjfys150,被用来运用flp/frt体系将烟曲霉bg盒插入在cbh1基因座处,插入频率为15.7%(或者为至少10.5%,因为其中一个菌株在摇瓶繁殖过程中显示出bg盒不稳定性)。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种检测水稻粒宽等位基因的特异性PCR分子标记和方法与流程
- 丝状真菌中多个基因拷贝的同时位点特异性整合的制造方法与工艺
- 一种特异性敲减人BRCC3基因慢病毒的构建及在肺癌中的应用的制造方法与工艺
- 烟粉虱MED隐种高温耐受相关基因特异性探针及其应用的制造方法与工艺
- CRISPR/SaCas9特异性敲除人CXCR4基因的方法与流程
- 玉米低镁条件下特异性转运镁离子基因ZmMGT10及其应用的制造方法与工艺
- CRISPR‑Cas9特异性敲除猪SLA‑3基因的方法及用于特异性靶向SLA‑3基因的sgRNA与制造工艺
- 构建在海马体区域特异性敲除IKKα基因的小鼠模型的方法及打靶载体和试剂盒与制造工艺
- 使用重叠非等位基因特异性引物和等位基因特异性阻断剂寡核苷酸的组合物进行等位基因特异性扩增的制造方法与工艺
- 用于检测烟草对tmv抗性的n基因特异性引物对、检测方法及试剂盒的制作方法