一种新型基因克隆T‑载体及其构建方法和应用与流程
本发明涉及基因工程领域,具体是涉及一种新型基因克隆T-载体及其构建方法和应用。
背景技术:
基因克隆为分子生物学实验的一个常规步骤,是基因功能和蛋白功能研究的基础。克隆PCR产物是基因克隆的主要部分,基因方法有多种,如T-载体法、PCR产物和载体均酶切的方法、USER cloning和ligase-independent cloning等。其中T-载体法由于其简便、快捷和高效等因素而最为常用。
T-载体克隆PCR产物以蓝白斑筛选来区分重组克隆和载体的自连。基于大肠杆菌半乳糖苷酶(LacZ)的α-互补蓝白斑筛选的基本原理如下。构建重组克隆的宿主菌如大肠杆菌DH10B和DH5α等基因组上的β-半乳糖苷酶N端31个氨基酸缺失,而克隆载体如pBluescript KS II(-)等的多克隆位点的一段核苷酸序列可编码如SEQ ID NO.9所示的含有相应的31个氨基酸的短肽(LQRRDWENPGVTQLNRLAAHPPFASWRNSEE)。pBluescript KS II(-)转化至大肠杆菌DH10B后,两者的半乳糖苷酶基因部分互相补充而形成具完整功能的β-半乳糖苷酶。在培养基中含有IPTG(异丙基硫代-β-D-半乳糖苷)和X-gal(5-溴-4氯-3-吲哚-β-D-半乳糖苷)时,IPTG诱导β-半乳糖苷酶高表达,酶切X-gal得到生色团而形成蓝色菌落。而当外源基因克隆至pBluescript KS II(-)的多克隆位点后,几乎不可避免地造成读码框的改变而不能表达能互补的31个氨基酸,进而不能获得有功能的β-半乳糖苷酶,X-gal不被酶切,含重组质粒的菌株不变色,呈现白色。挑取白斑来培养、提质粒、酶切和测序即可鉴定出重组克隆。
线性化的T-载体在其双链DNA的克隆位点分别含有5'-3'链的T碱基(此即T-载体的名称由来)以及3'-5'链的A碱基。由Taq扩增的PCR产物含有3'-A的突出端,在T4DNA连接酶的作用下,PCR产物和T-载体通过A-T碱基的相互连接而实现PCR产物的克隆。
常用的商业化T-载体为美国Promega公司的pGEM-T和pGEMT-easy以及日本Takara公司的pMD19-T和pMD19-T Simple。为保证片段的克隆,这些T-载体的5'-链的T碱基和3'-链的A碱基必需在连接后去除(由T4连接酶中的外切核酸酶活性)才能形成表达完整的31个氨基酸的短肽。以美国Promega公司的pGEM-T为例,其多克隆位点去除其中T和A碱基后,所表达的短肽为MTMITPSYLGDTIEYSSYASNALGALPYGRPAGGRTSDIPRPWRPGACDVGPNSPYSESYYNSLAVVLQRRDWENPGVTQLNRLAAHPPFASWRNSEEARTDRPSQQLRSLNGEWTRPVAAH.(“.”表示终止密码子)(SEQ ID NO.10),可互补宿主菌的β-半乳糖苷酶功能。去除T、A或同时去除T和A则不能获得功能化的短肽,菌株含有所得到的自连载体也表现为白斑,就不能和重组克隆区分开来。
这个克隆模式的必需基础是T4连接酶制品中含有外切核酸酶活性,未能实现和PCR产物连接的载体分子在由连接反应中外切核酸酶去除T和A而连接成为环状分子(含有T、A或T和A则不能连接)。
技术实现要素:
为克服目前T-载体依赖于T4连接酶制品中含有外切核酸酶活性这一局限性,
本发明提供了一种通过保留T和A碱基而制备T-载体和克隆PCR产物的方法。
具体的实验原理如下。将两侧含XcmI酶切位点的ccdB基因克隆至pBluescript II(KS-),分别获得pLS3417和pLS3424,克隆宿主菌为大肠杆菌DB3.1。pLS3417较pLS3424含有更多的酶切位点。XcmI酶切去除ccdB基因后获得T-载体。T-载体设计为通过保留T和A两个碱基而保持lacZα读码框结构,以提供α-互补的功能。以Taq聚合酶扩增的PCR产物和T-载体相连后,转化大肠杆菌克隆宿主菌DH10B,在含氨苄青霉素、异丙基硫代-β-D-半乳糖苷和5-溴-4氯-3-吲哚-β-D-半乳糖苷的筛选平板上,含有自连载体的菌株呈现篮斑,而含重组克隆的菌株为白斑,以此筛选得到重组克隆。以M13R和M13F为测序引物进行克隆片段的测序以验证克隆片段序列的正确性。
本发明公开了一种新型基因克隆T-载体,它通过下述方法构建得到:将序列如SEQ ID NO.13或SEQ ID NO.14的两侧含XcmI酶切位点的ccdB基因分别克隆至pBluescript II(KS-),分别获得pLS3417或pLS3424,宿主菌为大肠杆菌DB3.1;XcmI酶切去除ccdB基因后获得T-载体。
本发明还公开了上述T-载体的应用,是以Taq聚合酶扩增的PCR产物和T-载体相连后,转化大肠杆菌DH10B,在含氨苄青霉素、异丙基硫代-β-D-半乳糖苷和5-溴-4氯-3-吲哚-β-D-半乳糖苷的筛选平板上,含有自连载体的菌株呈现篮斑,而含重组克隆的菌株为白斑,以此筛选得到重组克隆。
由质粒pLS3417和pLS3424XcmI酶切而获得的T-载体采用不同于商业化
T-载体的克隆模式,读码框的选择,即克隆后保留T和A。载体pLS3417保留T和A后所表达的短肽为SEQ ID NO.11所示:MTMITPSAQLTLTKGNKSWVP
SLNSKDTAGCRDPSSNSPYSESYYARSLAVVLQRRDWENPGVTQLNRLAAHPPFASWRNSEEARTDRPSQQLRSLNGEWKL.;载体pLS3424保留T和A后所表达的短肽为SEQ ID NO.12所示:MTMITPSAQLTLTKGNKSWVPSKDTAGSS
NSPYSESYYARSLAVVLQRRDWENPGVTQLNRLAAHPPFASWRNSEEARTDRPSQQLRSLNGEWKL.。可见均含功能化的31个短肽序列。
质粒pLS3417和pLS3424的不同点在于前者的酶切位点多于后者,提供了更多的将所克隆的DNA片段切出重组克隆的选择;而pLS3424酶切位点较少,其优点是较少酶切位点的干扰。
高纯度和高质量中的T4连接酶中的外切核酸酶活性极低。随着未来T4连接酶产品质量的不断提高,外切核酸酶活性可能完全消失。如采用商业化T-载体的克隆模式,则T和A碱基不能去除,由此得到的含重组克隆或自连载体菌株均为蓝斑,这就影响了重组克隆的筛选。而采用本申请所提出的克隆模式就不存在这样的问题,得到的白斑的比例极大。
本专利申请的T-载体的另外一个特点是来源质粒(pLS3417和pLS424)中含有作用于DNA拓朴异构酶II的毒性基因ccdB。含ccdB基因的载体中需为宿主菌大肠杆菌DB3.1或其衍生菌株。这些菌株含DNA拓朴异构酶II的A亚单位的DNA螺旋酶GyrA A462C,即第462位精氨酸突变为半胱氨酸的突变,可抵抗CcdB的毒性。常规的克隆宿主菌不含有GyrA的A462C突变,含有ccdB基因的质粒将杀死菌株,而不能做为含ccdB基因的宿主菌。自含ccdB基因的质粒中制备T-载体的设计可避免或大幅避免微量的、未酶切的载体转化宿主菌而造成的转化背景干扰。
优选实施例中的两个PCR产物分别高效率地克隆至由pLS3417和pLS424所制备的T-载体,表明了本申请所得T-载体的适用性。本方法的优点包含:高效(高达90%),简洁,不收商业化载体的制约,可随时简便制备。
附图说明
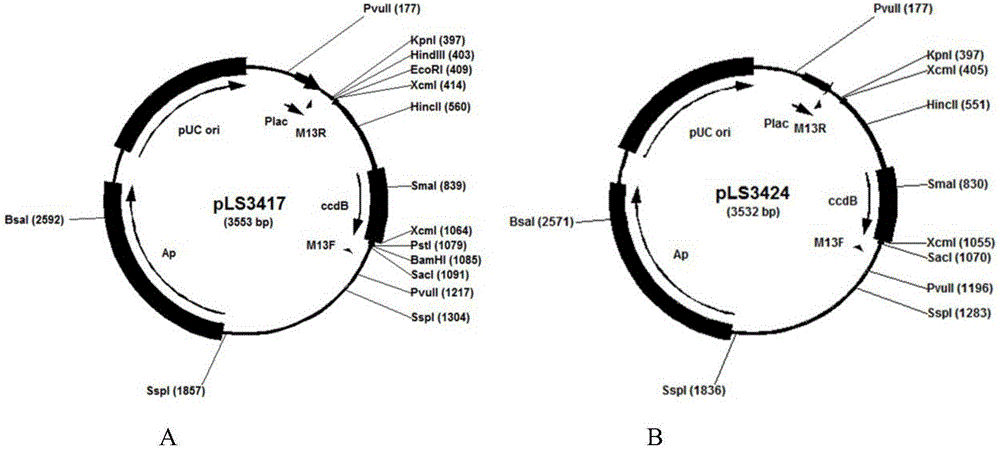
图1A.3553个碱基对的用于制备T-载体的质粒pLS3417的限制性酶切图谱。其中:
220-341是Plac启动子序列;
332-351是M13R测序引物序列;
759-1064是ccdB基因的开放阅读框序列;
1134-1150是M13F测序引物的反向互补序列;
1878-2738是氨苄青霉素抗性基因Ap的开放阅读框序列;
2886-3553是pUC复制子序列。
各基因的箭头方向为阅读框的方向;PvuII,KpnI,HindIII,EcoRI和XcmI等为限制性酶切位点,括号内的数字表示限制性酶切位点在质粒上的位置。
图1B.3532个碱基对的用于制备T-载体的质粒pLS3424的限制性酶切图谱。其中:
220-341是Plac启动子序列;
332-351是M13R测序引物序列;
750-1055是ccdB基因的开放阅读框序列;
1113-1129是M13F测序引物的反向互补序列;
1857-2717是氨苄青霉素抗性基因Ap的开放阅读框序列;
2865-3532是pUC复制子序列。
各基因的箭头方向为阅读框的方向;PvuII,KpnI和XcmI等为限制性酶切位点,括号内的数字表示限制性酶切位点在质粒上的位置。
图2A.由pLS3417制备的T-载体克隆2447bp PCR产物实验所得克隆提取质粒的凝胶电泳
1.质粒pBluescript II(KS-);
2.质粒pLS3417;
3-9.转化所得克隆培养所提取的质粒。
图2B.由pLS3417制备的T-载体克隆2447bp PCR产物所得到的5个正确大小的重组质粒的酶切结果
1.生工生物工程(上海)股份有限公司的DNA分子量标准SGM03(自上而下):
5.0,3.0,2.0,1.5,1.0,0.75,0.5,0.25,0.1kb;
2.一个正确大小的、未酶切的质粒;
3-7.5个正确大小的重组质粒的BamHI和HindIII双酶切。
图3A.由pLS3417制备的T-载体克隆3490bp PCR产物实验所得克隆提取质粒的凝胶电泳
1.质粒pBluescript II(KS-);
2.质粒pLS3417;
3-11.转化所得克隆培养所提取的质粒。
图3B.由pLS3417制备的T-载体克隆3490bp PCR产物所得到的6个正确大小的重组质粒的酶切结果
1.生工生物工程(上海)股份有限公司的DNA分子量标准SGM03(自上而下):
5.0,3.0,2.0,1.5,1.0,0.75,0.5,0.25,0.1kb;
2.一个正确大小的、未酶切的质粒;
3-8.6个正确大小的重组质粒的BamHI和HindIII双酶切。
图4A.由pLS3424制备的T-载体克隆2447bp PCR产物实验所得克隆提取质粒的凝胶电泳
1.质粒pLS3424;
2-11.转化所得克隆培养所提取的质粒。
图4B.由pLS3424制备的T-载体克隆2447bp PCR产物所得到的9个正确大小的重组质粒的酶切结果
1.生工生物工程(上海)股份有限公司的DNA分子量标准SGM03(自上而下):
5.0,3.0,2.0,1.5,1.0,0.75,0.5,0.25,0.1kb;
2.一个正确大小的、未酶切的质粒;
3-11.9个正确大小的重组质粒的KpnI和SacI双酶切。
图5A.由pLS3424制备的T-载体克隆3490bp PCR产物实验所得克隆提取质粒的凝胶电泳
1.质粒pLS3424;
2-9.转化所得克隆培养所提取的质粒。
图5B.由pLS3424制备的T-载体克隆3490bp PCR产物所得到的7个正确大小的重组质粒的酶切结果
1.生工生物工程(上海)股份有限公司的DNA分子量标准SGM03(自上而下):
5.0,3.0,2.0,1.5,1.0,0.75,0.5,0.25,0.1kb;
2.一个正确大小的、未酶切的质粒;
3-9.7个正确大小的重组质粒的KpnI和SacI双酶切。
具体实施方式
在本发明中所使用的术语,除非有另外说明,一般具有本领域普通技术人员通常理解的含义。
下面结合具体的实施例,并参照数据进一步详细地描述本发明。应理解,这些实施例只是为了举例说明本发明,而非以任何方式限制本发明的范围。
在以下的实施例中,未详细描述的各种过程和方法是本领域中公知的常规方法。所用试剂的来源、商品名以及有必要列出其组成成分者,均在首次出现时标明,其后所用相同试剂如无特殊说明,均以首次标明的内容相同。
实施例中所用到的菌株和质粒,均为已公开的菌株和质粒:
1.Escherichia coli MG1655。基因型F-LAM-rph-1,来自美国大肠杆菌保藏中心。
2.Escherichia coli DH10B。基因型F-mcrAΔ(mrr-hsdRMS-mcrBC)φ80dlacZΔM15ΔlacX74deoRrecA1araD139Δ(ara,leu)7697galUgalKrpsLendA1nupG。文献:Life Technologies,Inc.Focus,1990,12:19。购自美国Invitrogen公司。
3.Escherichia coli DB3.1,pMK2010。文献:House BL,Mortimer MW,Kahn ML.New Recombination Methods for Sinorhizobium meliloti Genetics.Applied Environmental Microbiology.2004,70(5):2806-2815.来自美国华盛顿大学Pullman分校Michael Kahn教授。
4.pBluescript II(KS-)。文献:Alting-Mees,M.A.and Short,J.M.(1989)pBluescript II:gene mapping vectors.Nucleic Acids Res,17,9494。基因克隆载体,购自美国Stratagene公司。
实施例
实施例1.制备T-载体所需质粒的构建
1.1 pLS3417的构建
设计引物C101:5'-GAAGGTACCAAGCTTGAATTCCAAGGATCTGCTGGGG
CTTACTAAAAGCCAGATAAC-3',(SEQ ID NO.1),下划线为引入的限制性酶切位点,斜体为XcmI酶切位点,其中EcoRI酶切位点的最后一个碱基C和XcmI酶切位点的第一个碱基C重合;C102:5'-GGAGAGCTCGGATCCCTGCAGC
CAGCAGTTCCTTGGTTATATTCCCCAGAACATCAGG-3,(SEQ ID NO.2),下划线为引入的限制性酶切位点,斜体为XcmI酶切位点)。XcmI的识别碱基序列为:5'-CCANNNNN↓NNNNTGG-5',其中N为任意的A,G,C,T中的任一碱基,↓表示酶切位点。本申请中将酶切位点前的一个碱基设计为T,且XcmI后去除的整个序列无终止密码子,在保留T和A碱基的情况下,自载体上的Plac启动子后的ATG开始,融合成一个读码框,且翻译后拥有互补β-半乳糖苷酶所需的多肽。
以pMK2010为模板,C101和C102PCR扩增得到0.7kb的两侧为酶切位点(含XcmI)、中间为ccdB基因的DNA片段SEQ ID NO.13。此0.7kb的DNA片段以KpnI和SacI酶切后与pBluescript II(KS-)相同酶切的载体片段相连,转化大肠杆菌DB3.1的感受态细胞,通过菌株培养,质粒提取,酶切和测序验证而筛选得到重组克隆pLS3417,其质粒物理图谱如图1A。
1.2 pLS3424的构建
设计引物C103:5'-GGAGGTACCATCCAAGGATCTGCTGGGGCTTACTAAA
AGCCAGATAAC-3',(SEQ ID NO.3),下划线为引入的限制性酶切位点,斜体为XcmI酶切位点;C104:5'-GGAGAGCTCCCAGCAGTTCCTTGGTTATATTCC
CCAGAACATCAGG-3',(SEQ ID NO.4),下划线为引入的限制性酶切位点,斜体为XcmI酶切位点)。如上,将XcmI酶切位点前的一个碱基设计为T,且XcmI后去除的整个序列无终止密码子,在保留T和A碱基的情况下,自载体上的Plac启动子后的ATG开始融合成一个读码框,且翻译后拥有互补β-半乳糖苷酶所需的多肽。
以pMK2010为模板,C103和C104PCR扩增得到0.7kb的两侧为酶切位点(含XcmI)中间为ccdB基因的DNA片段SEQ ID NO.14。此0.7kb的DNA片段以KpnI和SacI酶切后与pBluescript II(KS-)相同酶切的载体片段相连,转化大肠杆菌DB3.1的感受态细胞,通过菌株培养,质粒提取,酶切和测序验证而筛选得到重组克隆pLS3424,其质粒物理图谱如图1B。
实施例2.由pLS3417所制备的T-载体用于克隆PCR产物
2.1由pLS3417制备T-载体
pLS3417以XcmI(美国NEB公司)酶切得到0.7kb和2.9kb,回收2.9kb的载体片段,即得到克隆PCR产物所需的T-载体。
2.2 pLS3417制备T-载体用于克隆2447bp的PCR产物
设计引物C105:5'-GGATGAGCGGCATTTTCCGTG-3',(SEQ ID NO.5),C106:5'-GACCAACTGGTAATGGTAGC-3',(SEQ ID NO.6)。以大肠杆菌MG1655基因组DNA为模板,以Taq DNA聚合酶(美国NEB公司)PCR扩增得到2447bp的PCR产物(为lacZ基因的部分片段)。将此PCR产物凝胶回收后,设置连接反应体系为50ng的T-载体,50ng的PCR产物,1μl的T4DNA连接酶缓冲液(美国NEB公司)和1μl的T4DNA连接酶(美国NEB公司),总体积为10μl。16℃连接2小时后,转化大肠杆菌DH10B的化转感受态细胞,在含50μg/ml氨苄青霉素和20μg/ml IPTG+40μg/ml X-Gal的LB固体培养基上进行筛选。随机挑取7个白斑克隆进行培养,提质粒,琼脂糖凝胶电泳,以质粒pBluescript II(KS-)和质粒pLS3417为对照比较质粒大小,如图2A所示,5个克隆(第5至第9泳道)表现出正确的大小,另外2个克隆(第1和第2泳道)为异常连接。将5个大小正确的克隆以BamHI和HindIII双酶切,如图2B所示,均得到预期的2.5kb和2.9kb大小。随机选择其中1个克隆以M13R和M13F进行测序,发现序列与预期完全一致。这表明由pLS3417制备T-载体成功地克隆了2447bp的PCR产物,克隆效率达71%(5/7)。
2.3 pLS3417制备T-载体用于克隆3490bp的PCR产物
设计引物C107:5'-TGTTATATCCCGCCGTTAACC-3',(SEQ ID NO.7),C108:5'-TTCCTTACGCGAAATACGGGC-3',(SEQ ID NO.8)。同上以MG1655基因组DNA为模板,以Taq DNA聚合酶(美国NEB公司)PCR扩增得到3490bp的PCR产物(为lacZ基因的部分片段)。如上,回收DNA,连接转化。随机挑取9个白斑克隆培养,提质粒,琼脂糖凝胶电泳,以质粒pBluescript II(KS-)和质粒pLS3417为对照比较质粒大小,如图3A所示,6个克隆(第6至第11泳道)表现出正确的大小,另外3个克隆(第3至第5泳道)为异常连接。将这6个大小正确的克隆以BamHI和HindIII双酶切,如图3B所示,均得到预期2.9kb和3.5kb大小。随机选择其中1个克隆以M13R和M13F进行测序,发现序列与预期完全一致。这表明由pLS3417制备T-载体成功地克隆了3490bp的PCR产物,克隆效率达67%(6/9)。
实施例3.由pLS3424所制备的T-载体用于克隆PCR产物
3.1由pLS3424制备T-载体
pLS3424以XcmI(美国NEB公司)酶切得到0.7kb和2.9kb,回收2.9kb的载体片段,即得到克隆PCR产物所需的T-载体。
3.2 pLS3424制备T-载体用于克隆2447bp的PCR产物
同上以C105和C106为引物,大肠杆菌MG1655基因组DNA为模板,以Taq DNA聚合酶(美国NEB公司)PCR扩增得到相同2447bp的PCR产物。如上回收DNA,设置连接反应体系,转化大肠杆菌DH10B的化转感受态细胞进行克隆筛选。
随机挑取10个白斑克隆培养,提质粒,凝胶电泳,以质粒pLS3424为对照比较质粒大小,如图4A所示,9个克隆(第3至第11泳道)表现出正确的大小,另外1个(第2个泳道)克隆为异常连接。将这9个大小正确的克隆以KpnI和SacI双酶切,如图4B所示,均得到预期1.1,1.3和2.9kb大小。随机选择其中1个克隆以M13R和M13F进行测序,发现序列与预期完全一致。这表明由pLS3424制备T-载体成功地克隆了2447bp的PCR产物,克隆效率达90%(9/10)。
3.3 pLS3424制备T-载体用于克隆3490bp的PCR产物
同上以C107和C108为引物,大肠杆菌MG1655基因组DNA为模板,以Taq DNA聚合酶(美国NEB公司)PCR扩增得到相同3490bp的PCR产物。如上回收DNA,设置连接反应体系,转化大肠杆菌DH10B的化转感受态细胞进行克隆筛选。
随机挑取8个白斑克隆培养,提质粒,凝胶电泳,以质粒pLS3424为对照比较质粒大小,如图5A所示,7个克隆(第3至第9泳道)表现出正确的大小,另外1个克隆(第2泳道)为异常连接。将这7个大小正确的克隆以KpnI和SacI双酶切,如图5B所示,均得到预期1.2,2.3和2.9kb大小。随机选择其中1个克隆以M13R和M13F进行测序,发现序列与预期完全一致。这表明由pLS3424制备T-载体成功地克隆了3490bp的PCR产物,克隆效率达88%(7/8)。
SEQUENCE LISTING
<110> 南京师范大学
<120> 一种新型基因克隆T-载体及其构建方法和应用
<130>
<160> 14
<170> PatentIn version 3.3
<210> 1
<211> 57
<212> DNA
<213> 人工序列
<400> 1
gaaggtacca agcttgaatt ccaaggatct gctggggctt actaaaagcc agataac 57
<210> 2
<211> 58
<212> DNA
<213> 人工序列
<400> 2
ggagagctcg gatccctgca gccagcagtt ccttggttat attccccaga acatcagg 58
<210> 3
<211> 48
<212> DNA
<213> 人工序列
<400> 3
ggaggtacca tccaaggatc tgctggggct tactaaaagc cagataac 48
<210> 4
<211> 46
<212> DNA
<213> 人工序列
<400> 4
ggagagctcc cagcagttcc ttggttatat tccccagaac atcagg 46
<210> 5
<211> 21
<212> DNA
<213> 人工序列
<400> 5
ggatgagcgg cattttccgt g 21
<210> 6
<211> 20
<212> DNA
<213> 人工序列
<400> 6
gaccaactgg taatggtagc 20
<210> 7
<211> 21
<212> DNA
<213> 人工序列
<400> 7
tgttatatcc cgccgttaac c 21
<210> 8
<211> 21
<212> DNA
<213> 人工序列
<400> 8
ttccttacgc gaaatacggg c 21
<210> 9
<211> 31
<212> PRT
<213> 人工序列
<400> 9
Leu Gln Arg Arg Asp Trp Glu Asn Pro Gly Val Thr Gln Leu Asn Arg
1 5 10 15
Leu Ala Ala His Pro Pro Phe Ala Ser Trp Arg Asn Ser Glu Glu
20 25 30
<210> 10
<211> 122
<212> PRT
<213> 人工序列
<400> 10
Met Thr Met Ile Thr Pro Ser Tyr Leu Gly Asp Thr Ile Glu Tyr Ser
1 5 10 15
Ser Tyr Ala Ser Asn Ala Leu Gly Ala Leu Pro Tyr Gly Arg Pro Ala
20 25 30
Gly Gly Arg Thr Ser Asp Ile Pro Arg Pro Trp Arg Pro Gly Ala Cys
35 40 45
Asp Val Gly Pro Asn Ser Pro Tyr Ser Glu Ser Tyr Tyr Asn Ser Leu
50 55 60
Ala Val Val Leu Gln Arg Arg Asp Trp Glu Asn Pro Gly Val Thr Gln
65 70 75 80
Leu Asn Arg Leu Ala Ala His Pro Pro Phe Ala Ser Trp Arg Asn Ser
85 90 95
Glu Glu Ala Arg Thr Asp Arg Pro Ser Gln Gln Leu Arg Ser Leu Asn
100 105 110
Gly Glu Trp Thr Arg Pro Val Ala Ala His
115 120
<210> 11
<211> 102
<212> PRT
<213> 人工序列
<400> 11
Met Thr Met Ile Thr Pro Ser Ala Gln Leu Thr Leu Thr Lys Gly Asn
1 5 10 15
Lys Ser Trp Val Pro Ser Leu Asn Ser Lys Asp Thr Ala Gly Cys Arg
20 25 30
Asp Pro Ser Ser Asn Ser Pro Tyr Ser Glu Ser Tyr Tyr Ala Arg Ser
35 40 45
Leu Ala Val Val Leu Gln Arg Arg Asp Trp Glu Asn Pro Gly Val Thr
50 55 60
Gln Leu Asn Arg Leu Ala Ala His Pro Pro Phe Ala Ser Trp Arg Asn
65 70 75 80
Ser Glu Glu Ala Arg Thr Asp Arg Pro Ser Gln Gln Leu Arg Ser Leu
85 90 95
Asn Gly Glu Trp Lys Leu
100
<210> 12
<211> 95
<212> PRT
<213> 人工序列
<400> 12
Met Thr Met Ile Thr Pro Ser Ala Gln Leu Thr Leu Thr Lys Gly Asn
1 5 10 15
Lys Ser Trp Val Pro Ser Lys Asp Thr Ala Gly Ser Ser Asn Ser Pro
20 25 30
Tyr Ser Glu Ser Tyr Tyr Ala Arg Ser Leu Ala Val Val Leu Gln Arg
35 40 45
Arg Asp Trp Glu Asn Pro Gly Val Thr Gln Leu Asn Arg Leu Ala Ala
50 55 60
His Pro Pro Phe Ala Ser Trp Arg Asn Ser Glu Glu Ala Arg Thr Asp
65 70 75 80
Arg Pro Ser Gln Gln Leu Arg Ser Leu Asn Gly Glu Trp Lys Leu
85 90 95
<210> 13
<211> 701
<212> DNA
<213> 人工序列
<400> 13
gggtaccaag cttgaattcc aaggatctgc tggggcttac taaaagccag ataacagtat 60
gcgtatttgc gcgctgattt ttgcggtata agaatatata ctgatatgta tacccgaagt 120
atgtcaaaaa gaggtatgct atgaagcagc gtattacagt gacagttgac agcgacagct 180
atcagttgct caaggcatat atgatgtcaa tatctccggt ctggtaagca caaccatgca 240
gaatgaagcc cgtcgtctgc gtgccgaacg ctggaaagcg gaaaatcagg aagggatggc 300
tgaggtcgcc cggtttattg aaatgaacgg ctcttttgct gacgagaaca ggggctggtg 360
aaatgcagtt taaggtttac acctataaaa gagagagccg ttatcgtctg tttgtggatg 420
tacagagtga tattattgac acgcccgggc gacggatggt gatccccctg gccagtgcac 480
gtctgctgtc agataaagtc tcccgtgaac tttacccggt ggtgcatatc ggggatgaaa 540
gctggcgcat gatgaccacc gatatggcca gtgtgccggt ctccgttatc ggggaagaag 600
tggctgatct cagccaccgc gaaaatgaca tcaaaaacgc cattaacctg atgttctggg 660
gaatataacc aaggaactgc tggctgcagg gatccgagct c 701
<210> 14
<211> 679
<212> DNA
<213> 人工序列
<400> 14
ggtaccatcc aaggatctgc tggggcttac taaaagccag ataacagtat gcgtatttgc 60
gcgctgattt ttgcggtata agaatatata ctgatatgta tacccgaagt atgtcaaaaa 120
gaggtatgct atgaagcagc gtattacagt gacagttgac agcgacagct atcagttgct 180
caaggcatat atgatgtcaa tatctccggt ctggtaagca caaccatgca gaatgaagcc 240
cgtcgtctgc gtgccgaacg ctggaaagcg gaaaatcagg aagggatggc tgaggtcgcc 300
cggtttattg aaatgaacgg ctcttttgct gacgagaaca ggggctggtg aaatgcagtt 360
taaggtttac acctataaaa gagagagccg ttatcgtctg tttgtggatg tacagagtga 420
tattattgac acgcccgggc gacggatggt gatccccctg gccagtgcac gtctgctgtc 480
agataaagtc tcccgtgaac tttacccggt ggtgcatatc ggggatgaaa gctggcgcat 540
gatgaccacc gatatggcca gtgtgccggt ctccgttatc ggggaagaag tggctgatct 600
cagccaccgc gaaaatgaca tcaaaaacgc cattaacctg atgttctggg gaatataacc 660
aaggaactgc tgggagctc 679
- 还没有人留言评论。精彩留言会获得点赞!