一种3,6‑二去氧‑3‑氨基‑L‑艾杜糖及其衍生物的合成方法与流程
本发明属于化学合成领域,具体涉及一种3,6-二去氧-3-氨基-L-艾杜糖及其衍生物的合成方法。
背景技术:
氨基糖是脱氧糖中的一类最为重要的修饰糖,在化学、生物化学、医学以及药学等领域应用非常广泛。通常可分为两类,一类是指单糖分子中的一个或多个羟基被氮原子取代,另一类是指含有氮杂原子的糖类衍生物。氨基糖分子中的氨基经质子化后可产生带正电荷的铵离子,若将氨基酰化可适当降低其碱性与极性。氨基糖的这种结构特征,使其与其它具有生物活性的分子形成复合物后,可改善或提高其在临床上的疗效。氨基糖是一类具有重要生物活性的糖类衍生物的核心结构单元,广泛存在于生物体中,对许多生命活动的正常运行发挥着非常重要的作用。如2-氨基-2-去氧-D-吡喃葡萄糖在自然界中的分布很广,它的乙酰化衍生物存在于几丁质多糖中;2-氨基-2-去氧-D-半乳糖也大量存在,它是动物组织、软骨、硫酸软骨素和皮质素中多糖的重要结构单元。2-乙酰氨基-2-去氧-D-吡喃葡萄糖和D-半乳糖也以糖复合物(如糖脂类、脂多糖类、糖蛋白类)和粘多糖(如肝素、硫酸肝素、硫酸皮肤素、硫酸软骨素、透明质酸)以及寡糖类的形式广泛分布于生物体中[11]。氨基除了在C-2上,在C-3,C-4,C-5,C-6位上的氨基糖也大量存在。如唾液酸是5-氨基-5-去氧糖的九糖的衍生物,带负电荷,存在于细胞膜的表面,是细胞膜上负电荷的重要来源,与细胞的粘附密切相关。
我们要合成的3,6-二去氧-3-氨基-L-艾杜糖及其衍生物属于3-氨基吡喃糖,它是于20世纪90年代发现的一类大环内酰胺类新型抗生素的糖基部分。目前已用于临床上使用的许多蒽环类抗生素中就包含经结构修饰的各种3-氨基吡喃糖。如3-氨基-2,3,6-三去氧-L-来苏糖是天然存在的蒽环类正定霉素、阿霉素和洋红霉素的糖基部分。近年来发现的一些氮杂糖类药物作为糖苷酶抑制剂对糖尿病、肥胖症以及一些传染性疾病(如HIV)具有一定的疗效。总之,氨基糖普遍分布在一系列药物中,它在其中的作用是不可替代的,合成氨基糖及其衍生物也是当今糖化学工作者研究的热点课题之一。
技术实现要素:
本发明的目的在于提供一种新型、简捷、高效的合成3,6-二去氧-3-氨基-L-艾杜糖及其衍生物的方法,避免使用毒性大的重金属试剂。
本发明是通过以下技术方案实现的:
一种结构如下式10所示的3,6-二去氧-3-氨基-L-艾杜糖及其衍生物的合成方法,其合成路线如下:
其中,R1为H或甲基、乙基、烯丙基等常用的糖端基保护基;R2和R3分别独立地为H或乙酰基(-COCH3)、对甲苯磺酰基等常用的糖羟基上的酰基保护基,R4为H或苄基、烯丙基等常用的糖羟基上的醚类保护基。
进一步地,式(10)所示的化合物包括以下结构的化合物:
具体地,上述合成方法包括以下步骤:
1)以L-鼠李糖、乙酰氯和R1OH为原料,经亲核取代反应制得化合物1;
优选地,R1可为甲基、乙基或烯丙基;具体地,R1OH为甲醇;
2)以化合物1、原乙酸三甲酯、樟脑磺酸和无水二氯甲烷为原料,经取代反应制得化合物2;
3)以化合物2、硫磺三氧吡啶络合物、无水二甲基亚砜、三乙胺和无水二氯甲烷为原料,经氧化反应制得化合物3;
4)以化合物3、硼氢化钠和甲醇为原料,经还原反应制得化合物4;
5)以化合物4和R4Br为原料,经亲核取代反应制得化合物5;
优选地,R4为苄基或烯丙基;具体地,R4Br为苄基溴;
6)以化合物5、乙酸和N,N-二甲基甲酰胺为原料,经水解反应制得化合物6;
7)以化合物6、PCC、无水乙酸钠和二氯甲烷为原料,经氧化反应制得化合物7;
8)以化合物7、碳酸氢钠、羟胺盐酸盐和甲醇为原料,经脱水缩合反应制得化合物8;
9)以化合物8和冰乙酸为原料,以钯-碳为催化剂,经催化氢化反应制得化合物9;
10)以化合物9、吡啶和乙酸酐为原料,经乙酰化反应制得化合物10。
更具体地,上述合成方法包括以下步骤:
1)称取L-鼠李糖置于干燥的烧瓶中,取甲醇(试剂级)将其溶解,向溶液中滴加乙酰氯,加热至回流,反应完毕(一般12h后),反应液冷却至室温;冰浴下加入碳酸氢钠粉末中和,过滤,浓缩;用乙酸乙酯-甲醇体系柱分离,得到化合物1;
2)称取化合物1、樟脑磺酸放于烧瓶中,氩气氛围中注入二氯甲烷,冰浴下逐滴注入原乙酸三甲酯,原料逐渐溶解,反应30min;用二氯甲烷将反应液稀释,倒入饱和碳酸氢钠水溶液中,再用二氯甲烷萃取三次,合并有机相用无水MgSO4干燥,浓缩;用乙酸乙酯-石油醚体系柱分离,得到化合物2;
3)称取化合物2置于烧瓶中,用二氯甲烷将其溶解,并注入适量三乙胺,得到化合物2的溶液;另取一个烧瓶,称取硫磺三氧吡啶络合物并用无水二甲基亚砜溶解,冰浴下将该化合物2的溶液逐滴加入反应瓶中,冰浴下搅拌6h;反应液用乙醚稀释,蒸馏水洗涤,饱和硫酸铜溶液洗涤,饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩;用乙酸乙酯-石油醚体系柱分离得到化合物3;
4)称取化合物3置于烧瓶中,用试剂级的甲醇溶解,并置于冰浴中,称取适量硼氢化钠加入其中,反应完全(一般反应30min后),浓缩,用二氯甲烷稀释,将其倾入水中,再用二氯甲烷萃取,合并有机相,饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩;用乙酸乙酯-石油醚体系柱分离,得到化合物4;
5)称取化合物4、氢化钠和四丁基碘化铵置于烧瓶中,氩气氛围中注入无水N,N-二甲基甲酰胺将其溶解,再注入适量苄溴,室温搅拌过夜;反应液用乙醚稀释,后用蒸馏水洗涤,饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩;用乙酸乙酯-石油醚体系柱分离得到化合物5;
6)称取化合物5置于烧瓶中,注入无水N,N-二甲基甲酰胺将其溶解,冰浴下向其中加入乙酸水溶液,搅拌2h后TLC检测已水解完全,加入乙醚稀释,倾入冰的饱和碳酸氢钠水溶液中,中和至pH值为中性,乙醚萃取,合并有机相再用饱和氯化钠溶液反萃两次,无水MgSO4干燥,浓缩;用乙酸乙酯-石油醚体系柱分离得到化合物6;
7)称取化合物6置于烧瓶中,用二氯甲烷溶解,得化合物6的溶液;另取一个干燥的烧瓶作为反应瓶,并称取适量的PCC、无水乙酸钠和3 AMS放入其中;氩气氛围中注入无水二氯甲烷,冰浴下将化合物6的溶液加入其中,室温搅拌4h后反应完全,加入适量的硅胶粉末拌样浓缩至粉末状,用硅藻土装柱,乙醚淋洗,浓缩至剩余溶液(60mL)时,将其倾入饱和碳酸氢钠溶液中和至中性,用乙醚萃取,合并有机相再用饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩;用乙酸乙酯-石油醚体系柱分离得到化合物7;
8)称取化合物7置于烧瓶中,用试剂级的甲醇溶解,加入碳酸氢钠粉末,在冰浴下加入羟胺盐酸盐,搅拌20min后撤离冰浴室温搅拌,1h后TLC检测反应完全;用二氯甲烷稀释后倾入饱和碳酸氢钠溶液,二氯甲烷萃取三次,合并有机相并用饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩;用乙酸乙酯-石油醚体系柱分离得到化合物8;
9)称取化合物8置于烧瓶中,用冰乙酸将其溶解转移至反应釜中,称取催化量钯碳放入其中,于氢气氛中加压至550psi,升温至55℃,磁力搅拌,反应60h后停止反应,TLC检测原料反应完全;用硅藻土过滤,乙醇淋洗,浓缩得到化合物9,将其直接用于下一步反应;
10)称取化合物9置于烧瓶中,分别加入适量吡啶和乙酸酐室温搅拌,6h后TLC检测原料已反应完全;反应液用乙酸乙酯稀释,倾入冰的饱和碳酸氢钠溶液中,乙酸乙酯萃取,合并有机相,用饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩;用乙酸乙酯-石油醚体系柱分离得到化合物10;
上述合成方法中,
步骤1)中,L-鼠李糖、乙酰氯和甲醇的摩尔比为1:(0.05~0.1):(30~50);
步骤2)中,化合物1、原乙酸三甲酯、樟脑磺酸和无水二氯甲烷的摩尔比为:1:(1~3):(0.03~0.1):(10~30);
步骤3)中,化合物2、硫磺三氧吡啶络合物、无水二甲基亚砜、三乙胺和无水二氯甲烷的摩尔比为:1:(2~5):(10~20):(5~10):(30~50);
步骤4)中,化合物3、硼氢化钠和甲醇的摩尔比为:1:(0.5~2):(10~30);
步骤5)中,化合物4、苄基溴、氢化钠、四丁基碘化铵和N,N-二甲基甲酰胺的摩尔比为:1:(1~2):(1~2):(0.05~0.1):(30~50);
步骤6)中,化合物5、乙酸(80%水溶液)和N,N-二甲基甲酰胺的摩尔比为:1:(10~30):(30~50);
步骤7)中,化合物6、PCC、无水乙酸钠和二氯甲烷的摩尔比为:1:(1~3):(2~5):(30~50);
步骤8)中,化合物7、碳酸氢钠、羟胺盐酸盐和甲醇的摩尔比为:1:(1~3):(1~3):(30~50);
步骤9)中,化合物8、钯-碳催化剂和冰乙酸的摩尔比为:1:(0.1~0.3):(30~50);
步骤10)中,化合物9、吡啶和乙酸酐的摩尔比为:1:(5~15)(30~50)。
步骤1)柱分离所用乙酸乙酯-甲醇体系中乙酸乙酯与甲醇的体积比为60:1;
步骤2)柱分离所用乙酸乙酯-石油醚体系中乙酸乙酯与石油醚的体积比为1:3;
步骤3)柱分离所用乙酸乙酯-石油醚体系中乙酸乙酯与石油醚的体积比为1:6;
步骤4)柱分离所用乙酸乙酯-石油醚体系中乙酸乙酯与石油醚的体积比为1:4;
步骤5)柱分离所用乙酸乙酯-石油醚体系中乙酸乙酯与石油醚的体积比为1:1;
步骤6)柱分离所用乙酸乙酯-石油醚体系中乙酸乙酯与石油醚的体积比为1:(4~3);
步骤7)柱分离所用乙酸乙酯-石油醚体系中乙酸乙酯与石油醚的体积比为1:6;
步骤8)柱分离所用乙酸乙酯-石油醚体系中乙酸乙酯与石油醚的体积比为1:2;
步骤10)柱分离所用乙酸乙酯-石油醚体系中乙酸乙酯与石油醚的体积比为1:2。
本发明还提供下式11所示的化合物的合成方法,其与式10所示的化合物的合成方法区别仅在于步骤10)不同;式11所示的化合物的合成方法其步骤10)包括称取化合物9、氢化钠和四丁基碘化铵置于烧瓶中,氩气氛围中注入无水N,N-二甲基甲酰胺将其溶解,再注入适量苄溴,室温搅拌过夜。反应液用乙醚稀释,后用蒸馏水洗涤,饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩;用乙酸乙酯-石油醚体系柱分离得到化合物11。
其中,优选地,化合物9、苄基溴、氢化钠、四丁基碘化铵和N,N-二甲基甲酰胺的摩尔比为:1:(4~6):(6~10):(0.05~0.1):(50~80)。
本发明所述3,6-二去氧-3-氨基-L-艾杜糖衍生物与3,6-二去氧-3-氨基-L-艾杜糖具有相似的功能。
与现有技术相比,本发明具有以下有益效果:
本发明合成的3,6-二去氧-3-氨基-L-艾杜糖及其衍生物属于3-氨基吡喃糖,它是于20世纪90年代发现的一类大环内酰胺类新型抗生素的糖基部分。目前已用于临床上使用的许多蒽环类抗生素中就包含经结构修饰的各种3-氨基吡喃糖。如3-氨基-2,3,6-三去氧-L-来苏糖是天然存在的蒽环类正定霉素、阿霉素和洋红霉素的糖基部分。近年来发现的一些氮杂糖类药物作为糖苷酶抑制剂对糖尿病、肥胖症以及一些传染性疾病(如HIV)具有一定的疗效。
本发明的合成方法与文献已报道的相比较,操作简单,总收率相对较高,可以避免使用毒性很大的金属,如OsO4等,而且在最后的还原步骤中,可以采用不同的还原方法分别得到不同的氨基糖。
附图说明
图1、图2分别为化合物1的1H NMR图,13C NMR图。
图3为化合物2的1H NMR图。
图4为化合物4的1H NMR图。
图5为化合物5的1H NMR图。
图6、图7分别为化合物6的1H NMR图,13C NMR图。
图8为化合物7的1H NMR图。
图9、图10分别为化合物8的1H NMR图,13C NMR图。
图11为化合物10的1H NMR图。
图12为化合物14的1H NMR图。
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购买得到的常规产品。
实施例1
本实施例提供下式10所示化合物的合成方法,包括以下步骤:
(1)合成化合物1
称取L-鼠李糖(6.10g,37.16mmol)置于干燥的烧瓶中,取甲醇(试剂级)50mL将其溶解,向溶液中滴加乙酰氯(0.25mL,3.72mmol),加热至回流,反应6~12h后,反应液冷却至室温。冰浴下加入碳酸氢钠粉末中和,过滤,浓缩。用乙酸乙酯-甲醇体系(淋洗剂:乙酸乙酯/甲醇=60/1)柱分离,得到化合物1,产率95%。其反应方程式如下:
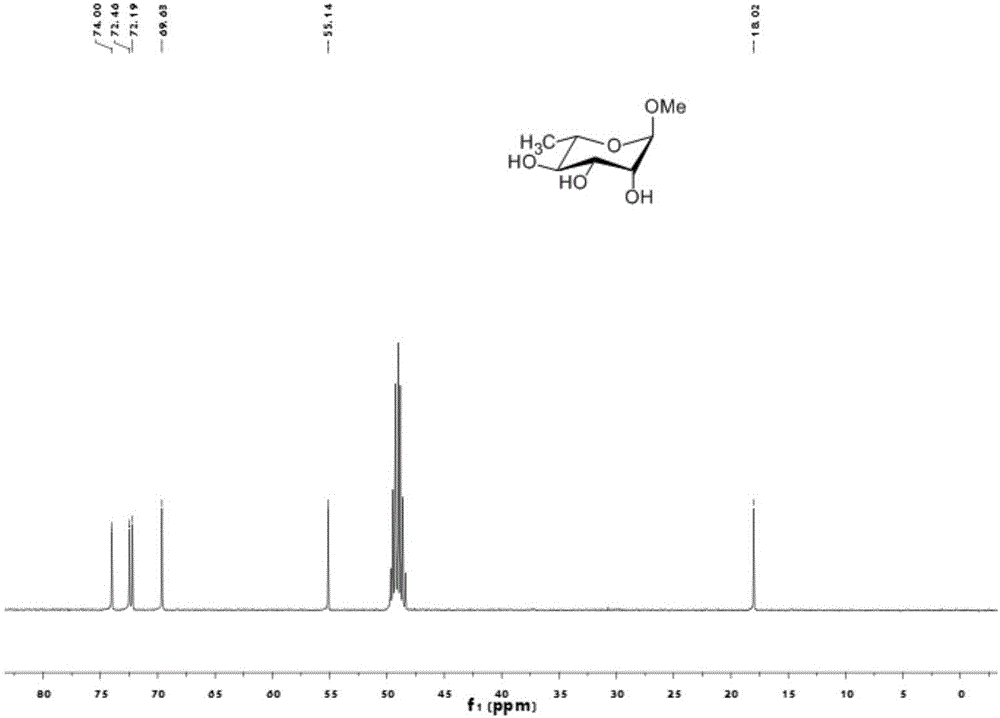
化合物1的1H NMR(核磁共振氢谱)见图1,13C NMR(核磁共振碳谱)见图2。
化合物1的结构表征:1H NMR(400MHz,MeOD)δ4.55(d,J=1.2Hz,1H),3.77(dd,J=1.6Hz,1H),3.52(m,1H),3.34(s,OMe),3.31-3.30(m,2H),1.26(d,J=6.4,3H).
13C NMR(100MHz,MeOD)δ102.81(C-1),74.00(C-5),72.46(C-2),72.19(C-4),69.63(C-3),55.14(OMe),18.02(C-6).
(2)合成化合物2
称取化合物1(677mg,3.80mmol)、樟脑磺酸(44mg,0.19mmol)放于烧瓶中,氩气氛围中注入二氯甲烷,冰浴下逐滴注入原乙酸三甲酯(1.0mL,7.86mmol),原料逐渐溶解,反应30min。用二氯甲烷将反应液稀释,倒入饱和碳酸氢钠水溶液中,再用二氯甲烷萃取三次,合并有机相用无水MgSO4干燥,浓缩。用乙酸乙酯-石油醚体系柱分离(淋洗剂:乙酸乙酯/石油醚=1/3),得到化合物2,产率97%。。其反应方程式如下:
化合物2的1H NMR见图3。
化合物2的结构表征:1H NMR(400MHz,CD3COCD3)δ4.82(d,J=2.4Hz,H-1),4.42(dd,J=7.2,4.8Hz,1H),3.98(dd,J=7.2,2.4Hz,1H),3.83(m,1H),3.79(dd,J=4.8,2.8Hz,1H),3.39(s,OMe),3.37(s,OMe),1.50(s,3H,CH3COMe),1.27(d,J=6.4Hz,H-6).
(3)合成化合物3
称取化合物2(2.11g,9.01mmol)置于烧瓶中,用二氯甲烷将其溶解,并注入适量三乙胺(6.3mL,45.20mmol),得到化合物2的溶液。另取一个烧瓶,称取硫磺三氧吡啶络合物(3.58g,22.47mmol)并用无水二甲基亚砜溶解,冰浴下将化合物2的溶液逐滴加入反应瓶中,冰浴下搅拌6h。反应液用乙醚稀释,蒸馏水洗涤,饱和硫酸铜溶液洗涤,饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩。用乙酸乙酯-石油醚体系柱分离(淋洗剂:乙酸乙酯/石油醚=1/6),得到化合物3,产率76%。其反应方程式如下:
(4)合成化合物4
称取化合物3(957.8mg,4.12mmol)置于烧瓶中,用试剂级的甲醇溶解,并置于冰浴中,称取适量硼氢化钠(156mg,4.12mmol)加入其中,30min后反应完全,浓缩,用二氯甲烷稀释,将其倾入水中,再用二氯甲烷萃取,合并有机相,饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩。用乙酸乙酯-石油醚体系柱分离(淋洗剂:乙酸乙酯/石油醚=1/4),得到化合物4,收率96%。其反应方程式如下:
化合物4的1H NMR见图4。
化合物4的结构表征:1H NMR(400MHz,CD3COCD3)δ4.80(d,1.6Hz,H-1),4.36(dd,J=6.8,4.4Hz,1H),4.08(dd,J=6.8,1.6Hz,1H),3.85(qd,J=6.5,2.1Hz,1H),3.72-3.67(m,1H),3.36(s,OMe),3.23(s,OMe),1.59(s,3H,CH3COMe),1.26(J=6.8Hz,H-6).
(5)合成化合物5
称取化合物4(70.6mg,0.3mmol)、氢化钠(25mg,0.63mmol)和四丁基碘化铵(5mg,0.013mmol)置于烧瓶中,氩气氛围中注入无水N,N-二甲基甲酰胺将其溶解,再注入适量苄溴(54μL,0.45mmol),室温搅拌过夜。反应液用乙醚稀释,后用蒸馏水洗涤,饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩。用乙酸乙酯-石油醚体系柱分离(淋洗剂:乙酸乙酯/石油醚=1/6),得到化合物5,产率97%。其反应方程式如下:
化合物5的1H NMR见图5。
化合物5的结构表征:1H NMR(400MHz,CDCl3)δ7.36-729(5H,Ph),4.74,4.60(d,J=12Hz,2H,CH2Ph),4.70(dd,J=6.4,3.6Hz,1H),4.40(d,J=4.8Hz,1H),4.12(m,1H),3.85(dq,J=12.8,6.4Hz,1H),3.55(dd,J=9.6,3.2Hz,1H),3.47(s,OMe),3.31(s,OMe),1.67(s,3H),1.31(d,J=6Hz,H-6).
(6)合成化合物6
称取化合物5置于烧瓶中,注入无水N,N-二甲基甲酰胺将其溶解,冰浴下向其中加入乙酸水溶液(2mL,80%),搅拌2h后TLC检测已水解完全,加入乙醚稀释,倾入冰的饱和碳酸氢钠水溶液中,中和至pH值为中性,乙醚萃取,合并有机相再用饱和氯化钠溶液反萃两次,无水MgSO4干燥,浓缩。用乙酸乙酯-石油醚体系柱分离(淋洗剂:乙酸乙酯/石油醚=1/4,1/3),得到化合物6,产率75%。其反应方程式如下:
化合物6的1H NMR见图6,13C NMR见图7。
化合物6的结构表征:1H NMR(400MHz,CDCl3)δ7.41-7.27(5H,Ph),4.90(d,J=4Hz,H-2),4.83,4.64(d,2H,CH2Ph),4.75(s,H-1),4.00(dt,J=10.0,4.0Hz,H-3),3.94(dd,J=13.2,6.8Hz,H-5),3.55(d,J=3.2Hz,H-4),3.35(s,3H,OMe),2.59(d,J=10.4Hz,3-OH),2.10(s,COCH3),1.35(d,J=6.8Hz,H-6).
13C NMR(100MHz,CDCl3)δ171.28(COCH3),138.55(1C,Ph),128.42(2C,Ph),127.73(1C,Ph),127.50(2C,Ph),98.99(C-1),78.85(C-4),75.88(C-2),70.51(CH2Ph),66.19(C-5),66.06(C-3),55.07(OMe),21.10(COCH3),16.78(C-6).
(7)合成化合物7
称取化合物6(1.52g,4.90mmol)置于烧瓶中,用二氯甲烷溶解,得化合物6的溶液;另取一个干燥的烧瓶作为反应瓶,并称取适量的PCC(即氯铬酸吡啶嗡盐,4.23g,19.6mmol)、无水乙酸钠(2.41g,29.38mmol)和3 AMS(即3A分子筛,1.5g)放入其中。氩气氛围中注入无水二氯甲烷,冰浴下向其中加入化合物6的溶液,室温搅拌4h后反应完全,加入适量的硅胶粉末拌样浓缩至粉末状,用硅藻土装柱,乙醚淋洗,浓缩至剩余溶液(60mL)时,将其倾入饱和碳酸氢钠溶液中和至中性,用乙醚萃取,合并有机相再用饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩。用乙酸乙酯-石油醚体系柱分离(淋洗剂:乙酸乙酯/石油醚=1:6),得到化合物7,产率91%。其反应方程式如下:
化合物7的1H NMR见图8。
化合物7的结构表征:1H NMR(400MHz,CDCl3)δ7.35-7.28(5H,Ph),5.85(d,J=8Hz,H-2),5.12(d,J=4Hz,H-1),4.73,4.36(d,J=11.6Hz,CH2Ph),4.15(qd,J=1.2Hz,H-5),3.68(d,J=1.6Hz,H-4),3.41(s,OMe),2.23(s,3H,COCH3),1.36(d,J=6.8Hz,H-6).
13C NMR(100MHz,CDCl3)δ200.63(C-3),170.66(COCH3),137.49(1C,Ph),129.37(2C,Ph),129.28(2C,Ph),129.03(1C,Ph),101.40(C-1),84.31(C-4),75.24(1C),72.76(1C),69.62(1C),56.36(OMe),21.43(COCH3),16.38(C-6).
(8)合成化合物8
称取化合物7(20mg,0.065mmol)置于烧瓶中,用试剂级的甲醇溶解,加入碳酸氢钠粉末(9.8mg,0.12mmol),在冰浴下加入羟胺盐酸盐(6.8mg,0.098mmol),搅拌20min后撤离冰浴室温搅拌,1h后TLC检测反应完全。用二氯甲烷稀释后倾入饱和碳酸氢钠溶液,二氯甲烷萃取三次,合并有机相并用饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩。用乙酸乙酯-石油醚体系柱分离(淋洗剂:乙酸乙酯/石油醚=1/2),得到化合物8,产率72%。。其反应方程式如下:
化合物8的1H NMR见图9,13C NMR见图10。
化合物8的结构表征:1H NMR(400MHz,CDCl3)δ8.22(s,N-OH),7.38-7.28(m,5H,Ph),5.79(d,J=3.6Hz,H-2),4.94(d,J=3.6Hz,H-1),4.83(d,J=1.2Hz,H-4),4.00(dt,J=18.4,6.8Hz,H-5),3.42(s,OMe),2.18(s,COCH3),1.30(d,J=6.4Hz,H-6).
13C NMR(100MHz,CDCl3)δ169.83(COCH3),150.46(C=N),137.63(1C,Ph),128.29(2C,Ph),128.26(2C,Ph),127.82(1C,Ph),98.58(C-1),71.07(1C),69.52(1C),68.14(1C),66.88(1C),20.68(1C),15.88(1C).
(9)合成化合物9
称取化合物8(120mg,0.37mmol)置于烧瓶中,用冰乙酸将其溶解转移至反应釜中,称取催化量钯碳(10%,40mg,0.038mmol)放入其中,于氢气氛中加压至550psi,升温至55℃,磁力搅拌,反应60h后停止反应,TLC检测原料反应完全。用硅藻土过滤,乙醇淋洗,浓缩得到化合物9。其反应方程式如下:
(10)合成化合物10
称取化合物9(69.2mg,0.32mmol)置于烧瓶中,分别加入适量吡啶(1mL)和乙酸酐(0.2mL)室温搅拌,6h后TLC检测原料已反应完全。反应液用乙酸乙酯稀释,倾入冰的饱和碳酸氢钠溶液中,乙酸乙酯萃取,合并有机相,用饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩。用乙酸乙酯-石油醚体系柱分离(淋洗剂:乙酸乙酯/石油醚=1/2),得到化合物10,产率95%。其反应方程式如下:
化合物10的1H NMR见图11。
化合物10的结构表征:1H NMR(400MHz,CDCl3)δ6.81(br,d,J=10Hz,NH),5.16(t,J=4.4Hz,H-2),4.97(d,J=2.8Hz,H-4),4.87(d,J=3.2Hz,H-1),4.38(m,H-3),4.13(dd,J=11.2,6.8Hz,H-5),3.47(s,OMe),2.15(s,COCH3),2.09(s,COCH3),2.03(s,COCH3),1.13(d,J=6.8Hz,H-6).
13C NMR(100MHz,CDCl3)δ170.77,170.69,170.43(3C,COCH3),99.10(C-1),72.51(C-4),66.07(1C),62.32(1C),56.82(1C),49.45(1C),24.36(1C),21.73(1C),21.67(1C),16.79(1C).
实施例2
本实施例提供下式11所示化合物的合成方法,其与实施例1的区别仅在于步骤10)不同。
式11所示的化合物的合成方法其步骤10)包括称取化合物9、氢化钠和四丁基碘化铵置于烧瓶中,氩气氛围中注入无水N,N-二甲基甲酰胺将其溶解,再注入适量苄溴,室温搅拌过夜。反应液用乙醚稀释,后用蒸馏水洗涤,饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩;用乙酸乙酯-石油醚体系柱分离得到化合物11。其反应方程式如下:
实施例3
本实施例提供下式12所示化合物的合成方法,其与实施例1的区别仅在于步骤9)称取化合物8(63mg,0.19mmol)置于烧瓶中,用干燥甲苯共沸三次并抽干,抽空换氩三次,注入无水四氢呋喃(1mL)将其溶解。另取一个10mL烧瓶作为反应瓶,称取四氢铝锂(25.5mg,0.67mmol)置于其中,抽空换氩三次,注入无水四氢呋喃(0.5mL),在冰浴下将原料的四氢呋喃溶液注入其中,恢复至室温后升高温度至回流。反应液从无色逐渐变为黄色,4h后经TLC检测原料已反应完全,冷却至室温,向其中依次加入蒸馏水(30μL),氢氧化钠水溶液(15%,30μL),蒸馏水(90μL),搅拌20min,加入无水MgSO4,搅拌30min,用混合溶剂(乙酸乙酯/甲醇=10/1)淋洗过滤,浓缩,得到化合物12。
实施例4
本实施例提供下式13所示化合物的合成方法,其包括首先按实施例3的方法制备得到化合物12,
然后以式12所示化合物为原料,通过以下步骤制备:取化合物12置于反应瓶中,依次加入重蒸吡啶(1mL),苯甲酰氯(0.3mL),N,N-二甲氨基吡啶(5mg),室温搅拌,6h后经TLC检测反应完全。将反应液用乙酸乙酯(10mL)稀释,倾入冰的饱和碳酸氢钠水溶液中,乙酸乙酯萃取(10mL×3),合并有机相,饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩。柱色谱梯度分离(乙醚/石油醚=4/1,乙酸乙酯/石油醚=3/1),得糖浆状化合物13,产率75%,其反应方程式如下:
实施例5
本实施例参考实施例1中合成化合物10的方法,具体通过以下步骤制备:向化合物12中依次加入重蒸吡啶(1mL),乙酸酐(0.3mL),N,N-二甲氨基吡啶(5mg),室温搅拌,6h后经TLC检测反应完全。将反应液用乙酸乙酯(10mL)稀释,倾入冰的饱和碳酸氢钠水溶液中,乙酸乙酯萃取(10mL×3),合并有机相,饱和氯化钠溶液反萃,无水MgSO4干燥,浓缩。柱色谱梯度分离(乙醚/石油醚=4/1,乙酸乙酯/石油醚=3/1),得糖浆状化合物14,产率75%。其反应方程式如下:
化合物14的1H NMR见图12。
化合物14的结构表征:1H NMR(400MHz,CDCl3)δ7.41-7.32(m,5H,Ph),5.53(br,d,J=8.4Hz,NH),5.10(dd,J=11.2,3.2Hz,H-2),4.84,4.43(d,J=12Hz,2H,CH2Ph),4.78(d,J=3.6Hz,H-1),4.49(m,H-3),4.04(q,J=6.4Hz,H-5),3.68(d,J=2.8Hz,H-4),3.40(s,OMe),2.06(s,COCH3),1.64(s,NHCOCH3),1.28(d,J=6.4Hz,H-6).
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种3,6‑二去氧‑3‑氨基‑L‑艾杜糖及其衍生物的合成方法与流程
- 取代2,4-(1H,3H)嘧啶二酮作为PARP抑制剂及其应用的制作方法与工艺
- 一种二氨基嘧啶化合物及包含该化合物的组合物的制作方法与工艺
- 2, 6‑二(2,4, 6‑三氨基‑5‑嘧啶偶氮)苯并(1, 2‑d; 4, 5‑d)双噻唑的制备方法和应用与流程
- 一种制备2-氨基-5,7-二甲氧基-三嗪并嘧啶的方法与流程
- 一种取代的二氨基嘧啶类化合物及包含该化合物的组合物及其用途的制作方法与工艺
- 一种2‑氨基‑4,6‑二甲氧基嘧啶制备的粗品后处理新工艺的制造方法与工艺
- 一种2‑氨基‑6‑甲氧基嘧啶‑4‑醇的制备工艺的制造方法与工艺
- 一种2‑甲氧基‑4‑氯‑5‑氟嘧啶的制备方法与流程
- 3‑甲氧基水杨醛缩‑5‑氨基‑1‑氢‑四氮唑席夫碱磁性配合物材料及合成方法与流程