双sgRNA文库的构建及其应用于高通量功能性筛选研究的方法与流程
本发明涉及基因编辑技术,具体涉及筛选功能性核酸序列,特别是功能性非编码元件或功能性成对基因的方法。
背景技术:
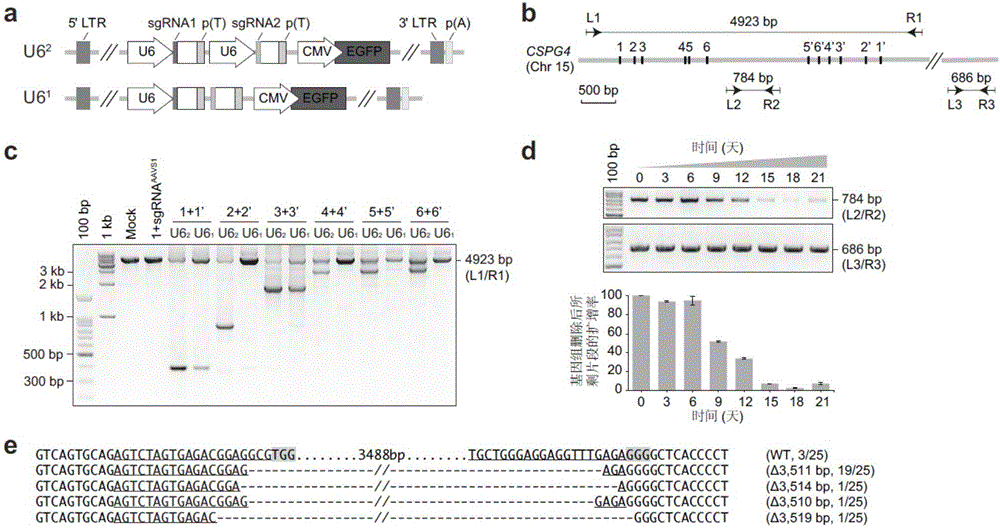
:来自细菌和古生菌的CRISPR/Cas系统[1]已被开发为基因组编辑工具,具有广泛的应用价值[2-4]。除了用于进行单个基因位点的编辑逐一研究基因的功能之外,也被报道开发出高通量功能性筛选方法,对编码基因的功能以及调节编码基因表达的关键增强子区域进行高通量筛选等[3-4]。在这些筛选中,可以通过将细胞生长或特定标志物作为选择信息,来选择与特定表型相关的单个向导RNAs(sgRNAs),从而研究其靶向基因或调节元件的功能[5-13]。除了很多单个的基因在重要的生物过程行使功能之外,很多生理过程是多基因多通路协同完成的,如何获得同时发挥某一重要功能的一组基因并进行高通量的筛选是一个没有解决的重要问题,这对于进一步研究基因的功能提出全新的模式具有重大意义。此外,哺乳动物基因组的绝大部分(约98%)由非编码区域组成,它们中的许多具有重要的调节作用。对非编码区域的功能分析一直是具有挑战性的,到目前为止还缺乏有效的高通量的筛选策略。虽然CRISPR/Cas系统在分析编码基因功能上具有广泛应用,但这种策略是基于破坏编码基因读码框的,对于非编码元件来说可能并不好用,对于非编码元件来说,由一个gRNA引起的插入缺失indels不太可能产生丧失功能的表型。虽然已有报道使用成对gRNA(pairedgRNA,pgRNA)来产生基因组的缺失,以研究单个lncRNA的功能[14,15],但用于鉴别功能性非编码元件的高通量策略还未见有报道。先前已报道了一种构建成对gRNACRISPR-Cas9文库的方法(JoanaA.Vidigaletal.,Rapidandefficientone-stepgenerationofpairedgRNACRISPR-Cas9libraries,NatureCommunications,2015),但该方法操作较为繁琐,不够简便,当用于大规模高通量筛选时仍然需要较大的工作量,且到目前为止并没有用该方法成功构建高通量成对gRNA文库的实例报道。本领域中仍然需要开发简便可行的高通量筛选策略,用于进行功能性核酸序列,例如功能性非编码元件或功能性成对基因的高通量筛选。技术实现要素:本发明提供构建pgRNA表达质粒文库的方法,所述pgRNA表达质粒文库当与Cas蛋白一同被引入细胞中时,可以使得基因组上两个sgRNA靶位点被切割,引起靶核酸序列的敲除,通过靶核酸序列的敲除筛选功能性的核酸序列。例如,本发明中,pgRNA可以靶向非编码元件,从而导致基因组非编码元件上两个sgRNA靶位点之间的区域发生缺失,引起靶非编码元件的敲除,从而进行功能性非编码元件的高通量筛选。再例如,本发明中,pgRNA含有的两个sgRNA还可以分别靶向两个在功能上有关联的不同基因,在细胞中同时引发双基因的敲除,从而对功能性成对基因进行高通量筛选。本发明还提供构建核酸序列,例如非编码元件(例如lncRNA基因)或成对基因基因敲除文库的方法,所述核酸序列,例如非编码元件(例如lncRNA基因)或成对基因敲除文库通过将本发明的pgRNA表达质粒文库转入细胞获得,所述核酸序列,例如非编码元件(例如lncRNA基因)或成对基因敲除文库可用于筛选功能性的核酸序列,例如功能性非编码元件(例如lncRNA基因)或功能性成对基因。本发明还提供筛选功能性核酸序列,例如功能性非编码元件(例如lncRNA基因)或功能性成对基因的方法。根据本发明的一个方面,提供构建pgRNA表达质粒文库的方法,包括:(1)提供初始质粒,所述初始质粒包含顺序连接的第一U6启动子、第二gRNA骨架序列编码序列和转录终止子;(2)通过第一步连接反应将多个“第一gRNA配对序列编码序列-间隔序列-第二gRNA配对序列编码序列”的每一个分别插入到初始质粒上的第一U6启动子和第二gRNA骨架序列编码序列之间,然后转化感受态细胞获得第二质粒混合物;(3)通过第二步连接反应将顺序连接的第一gRNA骨架序列、转录终止子和第二U6启动子插入到第二质粒中两个gRNA配对序列之间,然后转化感受态细胞获得pgRNA表达质粒文库。在一些实施方案中,所述第一步连接反应是将在第一U6启动子的3'端和第二gRNA骨架序列编码序列的5'端被切割的初始质粒与多个DNA寡核苷酸序列的混合物进行连接反应。在一些实施方案中,初始质粒被限制性核酸内切酶,优选II型(TypeIIs)限制性核酸内切酶,更优选BsmBI切割。在一些实施方案中,初始质粒中的第一U6启动子和第二gRNA骨架序列编码序列之间可以含有在其两端具有限制性核酸内切酶酶切位点的自杀基因。在优选的实施方案中,所述自杀基因为ccdB基因。优选地,所使用的限制性内切酶是II型(TypeIIs)限制性核酸内切酶。更优选地,所使用的限制性内切酶是BsmBI。在一些实施方案中,所述多个DNA寡核苷酸序列的每一个包含一个pgRNA的两个gRNA配对序列,即第一gRNA配对序列、第二gRNA配对序列的编码序列,并且包含顺序连接的“第一gRNA配对序列编码序列-间隔序列-第二gRNA配对序列编码序列”的序列。在一些实施方案中,所述DNA寡核苷酸序列的间隔序列中包含切割位点,以便于在第一gRNA配对序列编码序列和第二gRNA配对序列编码序列之间进行切割以进行第二步连接反应。优选地,所述切割位点是限制性核酸内切酶切割位点,优选II型(TypeIIs)限制性核酸内切酶切割位点,更优选BsmBI切割位点。在一些实施方案中,所述DNA寡核苷酸序列的两端还可以具有与引物配对的序列,以便于通过引物配对介导的扩增反应扩增所述DNA寡核苷酸序列。在一些实施方案中,在进行第一步连接反应之前,对所述DNA寡核苷酸序列混合物进行扩增。在一些实施方案中,第一步连接反应通过Gibson组装方法进行。在一些实施方案中,每一个第二质粒中在第一U6启动子和第一gRNA配对序列编码序列之间,以及第二gRNA配对序列编码序列和第二gRNA骨架序列编码序列之间是紧邻连接。在一些实施方案中,所述第二步连接反应是将在第一gRNA配对序列编码序列的3'端和第二gRNA配对序列编码序列的5'端被切割的第二质粒混合物与包含“第一gRNA骨架序列编码序列-转录终止子-第二U6启动子”序列的DNA片段进行连接反应。在一些实施方案中,第二质粒混合物被限制性核酸内切酶,优选II型(TypeIIs)限制性核酸内切酶,更优选BsmBI切割。在一些实施方案中,所述包含“第一gRNA骨架序列编码序列-转录终止子-第二U6启动子”序列的DNA片段中在转录终止子和第二U6启动子之间还包含接头序列,所述接头序列可以包含引物结合区域,用于扩增含有第一gRNA配对序列编码序列和/或第二gRNA配对序列编码序列的片段。在一些实施方案中,每一个pgRNA表达质粒中第一U6启动子和第一gRNA配对序列编码序列之间,以及第一gRNA配对序列和第一gRNA骨架序列编码序列之间是紧邻连接。在一些实施方案中,每一个pgRNA表达质粒中第二U6启动子和第二gRNA配对序列编码序列之间,以及第二gRNA配对序列和第二gRNA骨架序列编码序列之间是紧邻连接。在一些实施方案中,所述初始质粒是慢病毒载体质粒。在一些实施方案中,第一U6启动子和第二U6启动子可以是相同的或不同的。在一些实施方案中,第一gRNA骨架序列和第二gRNA骨架序列可以是相同的或不同的。在一些实施方案中,初始质粒还包含与启动子可操作相连的标记物基因。在一些实施方案中,所述标记物基因是抗生素抗性基因或荧光蛋白基因。在一些实施方案中,所述pgRNA靶向非编码元件。在一些实施方案中,所述pgRNA表达质粒文库靶向超过50个非编码元件,优选超过100个非编码元件、超过200个非编码元件,超过500个非编码元件,甚至超过1000个非编码元件。在一些实施方案中,所述pgRNA表达质粒文库中不同的pgRNA靶向不同的非编码元件,或者多个pgRNA靶向同一个非编码元件,或者是二者的组合。在一些实施方案中,所述非编码元件是非编码基因,优选lncRNA基因。在一些实施方案中,所述pgRNA靶向成对基因。即,pgRNA中所包含的两个gRNA分别靶向成对基因中的两个基因。在一些实施方案中,所述pgRNA表达质粒文库靶向超过50对成对基因,优选超过100对成对基因、超过200对成对基因,超过500对成对基因,甚至超过1000对成对基因,或者甚至超过10000对成对基因。在一些实施方案中,所述成对基因优选是成对编码基因。在一些实施方案中,第一gRNA配对序列或第二gRNA配对序列的靶位点独立地位于靶基因的启动子或基因本体上。在一些实施方案中,所述转录终止子是polyT。根据本发明的另一个方面,提供构建核酸序列敲除细胞文库的方法,包括:使用上述方法构建pgRNA表达质粒文库,将所述pgRNA表达质粒文库递送到靶细胞中,并向靶细胞引入Cas9核酸酶,培养细胞后筛选成功转入所述pgRNA表达质粒文库的细胞,获得核酸序列敲除细胞文库。在一些实施方案中,所述核酸序列是非编码元件或成对基因。在一些实施方案中,通过慢病毒感染将所述pgRNA表达质粒文库递送到靶细胞中。在一些实施方案中,将所述pgRNA表达质粒文库与病毒包装质粒共转染宿主细胞,产生含有所述pgRNA表达质粒文库的慢病毒,用该慢病毒感染靶细胞,从而将所述pgRNA表达质粒文库递送到靶细胞中。在一些实施方案中,慢病毒感染的MOI小于等于0.3。在优选的实施方案中,感染细胞到收集被感染细胞之间的培养时间为约48h至约72h。在优选的实施方案中,通过药物抗性或FACS方法收获受感染的细胞。根据本发明的另一个方面,提供通过上述方法构建获得的核酸序列敲除细胞文库。根据本发明的另一个方面,提供筛选功能性核酸序列的方法,包括:培养上述细胞文库或在特定的筛选条件下培养上述细胞文库,然后提取细胞文库混合物的基因组DNA,扩增包含pgRNA对中的任何一个或两个gRNA配对序列编码序列的DNA片段,利用深度测序技术对扩增产物进行测序,分析测序结果,从而确定pgRNA的靶核酸序列的功能。在一些实施方案中,所述核酸序列是非编码元件或成对基因。在一些实施方案中,所述培养的时间足以使所希望筛选的核酸序列的功能被检测到。在一些实施方案中,测序结果中与对照相比占比提高的pgRNA说明其靶核酸序列的敲除有利于细胞增殖,测序结果中与对照相比占比降低的pgRNA说明其靶核酸序列的敲除不利于细胞增殖。在一些实施方案中,所述非编码元件是lncRNA基因。在一些实施方案中,测序结果中与对照相比占比提高的pgRNA说明其靶非编码元件的敲除有利于细胞增殖,测序结果中与对照相比占比降低的pgRNA说明其靶非编码元件的敲除不利于细胞增殖。在一些实施方案中,所述成对基因是成对编码基因。在一些实施方案中,测序结果中与对照相比占比提高的pgRNA说明其靶成对基因的敲除有利于细胞增殖,测序结果中与对照相比占比降低的pgRNA说明其靶成对基因的敲除不利于细胞增殖。本发明是一种高通量CRISPR/Cas策略,可以使用多对gRNA(pgRNA)产生大量核酸序列的删除,使得能够进行基因组中核酸序列的高通量功能性研究。例如,本发明可以使用多对gRNA(pgRNA)产生大量长的非编码RNA(lncRNA)的大片段删除,使得能够进行哺乳动物非编码元件的高通量功能性研究。本发明还可以使用多对gRNA(pgRNA)产生成对基因的敲除,通过高通量的筛选获得并研究多个基因共同发挥重要功能的实例。本发明的方法还可以用于研究除简单生长以外的感兴趣的其它表型与功能,还可以更广泛地应用于研究其它非编码序列,包括microRNAs、顺式调控元件和其它功能未知的元件。附图说明图1是在稳定表达Cas9的人细胞中,慢病毒递送的pgRNA以高效率产生大片段删除。(a)表达成对向导RNA(pgRNA)的慢病毒质粒的结构。将U6启动子和gRNA编码序列克隆到LL3.7慢病毒骨架中。通过金门方法(GoldenGatemethod)将扩增的编码pgRNA的DNA片段连接到具有两个U6启动子(U62)或者只有一个共用的U6启动子(U61)的慢病毒骨架中。(b,c)pgRNA载体通过慢病毒被递送到表达Cas9的人细胞中。通过PCR鉴别了由靶向CSPG4基因的pgRNA诱导的大片段删除。选择六对产生~2-4.5kb的大片段删除的gRNA(b),并使用引物L1/R1进行基因组PCR反应(b,c)。通过FACS富集所有感染的Huh7.5OC细胞,并孵育6天。图1a中字母U62和U61代表两种串联方式,对照是一对gRNA,其中一个靶向CSPG4基因座,另一个靶向AAVS1区域。(d)细胞感染pgRNA病毒后,培养时间对大片段删除的效率的影响。pgRNA(图1c中的3+3’,设计的是产生~3.5kb的缺失)通过慢病毒感染被递送到Huh7.5OC细胞中,在所示的不同时间点(上图)提取基因组DNA,使用相应于pgRNA靶位点侧翼序列的引物L2/R2(b)进行定量,使用相应于远离pgRNA靶位点的序列的引物L3/R3(b)进行标准化。引物序列参见后面表12。使用ImageJ软件分析图像,数据表示为平均值±标准差(n=3)(下图)。(e)感染后3周,对混合细胞中pgRNA(3+3’)靶向的人CSPG4基因座中的大片段删除的DNA测序分析(图1d)。含有两个gRNA的靶区域的靶基因的部分序列以下划线表示,阴影核苷酸代表PAM序列。短横线表示删除。图2是由慢病毒介导的pgRNA诱导的大片段删除。(a,b)选择5对诱导1-5kb的大片段删除的gRNA,这些gRNA靶向MALAT1基因并具有各自的U6启动子(U62),使用引物L4/R4进行基因组PCR反应。通过FACS富集所有感染的Huh7.5OC细胞,并孵育6天。对照是一对gRNA,其中一个靶向MALAT1基因,另一个靶向AAVS1区域。(c)细胞感染靶向MALAT1基因的pgRNA病毒后,培养时间对大片段删除的效率的影响。pgRNA(b中的2+2’,按照设计会产生4.3kb的删除)通过慢病毒感染被递送到Huh7.5OC细胞中,在给定的不同时间点提取基因组DNA。使用相应于pgRNA靶位点侧翼序列的引物L5/R5进行定量。(d)细胞感染靶向CSPG4基因的pgRNA病毒后,培养时间对大片段删除的效率的影响。pgRNA(c中的2+2’,按照设计会产生4.0kb的删除)通过慢病毒感染被递送到Huh7.5OC细胞中,在给定的不同时间点提取基因组DNA。使用相应于pgRNA靶位点侧翼序列的引物L2/R2进行定量,使用相应于远离靶位点的序列的引物L3/R3进行标准化。所有引物序列列于表12中。通过ImageJ软件分析图像,数据表示为平均值±标准差(n=3)。(e)对感染后3周,混合细胞中由图1d中的两个CSPG4-靶向pgRNA(p2和p4,分别代表2+2’和4+4’),和图2c中的两个MALAT1-靶向pgRNA(p2和p3,分别代表2+2’和3+3’)诱导的大片段删除的DNA测序分析。基因组中含有两个gRNA的靶区域的靶基因的部分序列用下划线表示,阴影核苷酸代表PAM序列。短横线代表删除。图3是pgRNA文库的设计、克隆和筛选。(a)pgRNA质粒文库构建。每个合成的137-nt的DNA寡核苷酸含有两个gRNA配对序列编码序列。扩增寡聚物以产生dsDNA分子,并使用Gibson反应将其克隆到慢病毒骨架中。通过BsmBI消化和连接插入接头区段之后,获得最终的构建体(下文“材料与方法”部分)。(b)通过慢病毒感染将pgRNA文库递送到Huh7.5OC细胞中,MOI为大约0.3。感染后3天,通过FACS用绿色荧光收获感染的细胞。为进行筛选,在进行基因组DNA提取和高通量测序分析之前将文库细胞培养30天。图4是所设计的寡核苷酸和每一对的两个gRNA之间的接头的DNA序列,以及进行PCR扩增以进行深度测序分析,如电泳图所示。(a)每一个寡核苷酸的特征和序列。左臂和右臂用于扩增时的引物靶定。(b)pgRNA质粒构建的接头的特征,以及它在第一gRNA骨架的末端和第二U6启动子的起始端之间的独特序列。(c)从Huh7.5OC文库和野生型细胞分离的基因组DNA被用作PCR扩增的模板。图5是负向筛选(negativelyselected)和正向筛选(positivelyselected)的lncRNA的鉴别。(a)靶向阴性对照、阳性对照和lncRNA的pgRNA的丰度(logfold)变化分布。*P<0.05;**P<0.01;Wilcox秩和检验。中间的线代表中位数值;方框代表四分位差;每个触须(whiskers)延伸至四分位差的1.5倍;圆点代表离群值。(b)所有靶向一种阳性对照基因EZH2的pgRNA的对数倍数(logfold)和基因组位置总结。大部分pgRNA被减低(即转入该pgRNA的细胞减少),包括靶向EZH2的启动子、启动子+外显子和内含子的的pgRNA(logFC<-1)。(c,d)由MAGeCK计算获得的排名靠前的负向筛选lncRNA(c)和正向筛选lncRNA(d)的排序聚集(RobustRankAggregation,RRA)得分。被负向筛选的一些阳性对照基因也以黑色三角显示。更小的RRA得分表示对相应lncRNA的更强的选择。图6是独立实验重复的相关性和read分布。(a)对照样品的重复组之间的相关性。(b)30-天富集样品的重复组之间的相关性。还给出了重复之间的皮尔森相关系数(PearsonCorrelationCoefficient,PCC)。图7是靶向排名靠前的负向筛选lncRNA(a)和正向筛选lncRNA(b)的pgRNA的平均read计数。(c)靶向AAVS1(左)和非靶向对照(右)的pgRNA的平均read计数。图8是被选择用于验证的负向(a)和正向(b)筛选的lncRNA的pgRNAread计数。图9是候选lncRNA的验证。(a-c)Huh7.5OC细胞中RPL18A(a)、负向筛选的lncRNA(b)和正向筛选的lncRNA(c)的大片段删除对于细胞增殖的效果。每一个lncRNA选择3-5对靶向启动子或启动子+外显子的pgRNA进行验证。将pgRNA通过慢病毒感染递送到细胞中,并通过FACS对EGFP+的细胞进行定量。第一次定量是在病毒感染后三天开始,在本图以及之后的图中标记为第0天。用对照(第0天)对给定时间点的EGFP+百分比进行标准化,确定细胞增殖率。将新设计的与原始文库中所使用的不同的pgRNA用星号(*)标注。箭头指向转录起始位点。空心方框和阴影方框分别指非编码和编码基因的外显子。(d)负向筛选的lncRNA的转录抑制对于细胞增殖的效果。对RPL18A、AC004463.6、RP11-439K3.1和AC095067.1的mRNA水平(相对于GAPDH标准化)进行定量。定量PCR所使用的所有引物都在表13中列出。(e)Huh7.5OC细胞中靶向负向筛选的lncRNA的sgRNA和pgRNA对于细胞增殖的效果。数据表示为平均值±标准差(n=3)。通过Student’st检验计算P值,并用BenjaminiHochberg程序进行多重比较的校正,*P<0.05;**P<0.01;***P<0.001;NS:无显著性。图10是Huh7.5细胞中候选lncRNA的基因组验证和由RP11-439K3.1靶向pgRNA诱导的基因组缺失。(a)Huh7.5OC细胞中负向筛选的lncRNA的大片段删除对于细胞增殖的影响。(b)Huh7.5OC细胞中正向筛选的lncRNA的大片段删除对于细胞增殖的影响。(c)含有LINC00882-靶向pgRNA的Huh7.5OC细胞中的LINC00882mRNA水平,和回补LINC00882的cDNA后的Huh7.5OC细胞中的LINC00882mRNA水平(相对于GAPDH标准化)被定量。定量PCR所使用的所有引物列在表13中。(d)LINC00882-靶向pgRNA和LINC00882的cDNA回补对于Huh7.5OC细胞增殖的影响。数据表示为平均值±标准差(n=3)。使用Student’st检验计算P值,并用BenjaminiHochberg程序进行多重比较校正,**P<0.01。(e)RP11-439K3.1_p3和RP11-439K3.1_p4通过慢病毒被递送到Huh7.5OC细胞中,通过基因组PCR检测缺失。(f)Huh7.5OC细胞中靶向负向筛选的lncRNA的sgRNA(转录抑制)和pgRNA对细胞增殖的效果。细胞增殖的测定与图9中所述的相同。在本图中以及其余的图中,新设计的与原始文库中所使用的不同的gRNA对被标记上星号(*)。箭头表示转录起始位点。空心方框和阴影框分别指非编码和编码基因的外显子。数据表示为平均值±标准差(n=3)。使用Student’st检验计算P值,并用BenjaminiHochberg程序进行多重比较校正,*P<0.05;**P<0.01;***P<0.001;NS:不显著。图11是用所给出的pgRNA和转录抑制的sgRNA对Huh7.5细胞生存能力的影响。所有pgRNA和sgRNA通过慢病毒感染被递送到Huh7.5OC细胞中。感染后72h对细胞进行FACS富集,FACS后1-3天进行LDH致死性检测。数据表示为平均值±标准差(n=3)。图12是正向筛选lncRNA的转录激活对于细胞活力的效果。(a)对LINC01087和LINC00882mRNA的水平(相对于GAPDH标准化)进行定量。定量PCR所用的所有引物列在表13中。(b)所有sgRNA通过瞬时转染被递送到Huh7.5细胞中。感染后72h对细胞进行进行FACS富集,FACS后1-3天进行LDH致死性检测。数据表示为平均值±标准差(n=3)。图13是成对sgRNA(pgRNA)的全序列。具体实施方式本发明构建了靶向细胞中多个核酸序列,例如非编码元件(如lncRNA基因)或成对基因的pgRNA表达质粒文库,通过使用Cas9/CRISPR(ClusteredRegularlyInterspacedShortPalindromicRepeats,成簇的规律间隔的短回文重复序列)系统和所述pgRNA文库实现对细胞中靶核酸序列的敲除,从而通过表型特征筛选功能性核酸序列,例如功能性非编码靶基因(如功能性lncRNA基因)或功能性成对基因。除非根据上下文可明确其有另外的含义,本发明所使用的术语“核酸序列”或“靶核酸序列”可以指基因组中存在的任何具有已知功能或未知功能的核酸序列,例如但不限于非编码元件、成对基因、未知功能的核酸序列等。本发明中所述的“一个核酸序列”是指具有一种功能或可能具有一种功能的完整核酸序列。本发明中所述的“不同核酸序列”是指分别具有不同功能的核酸序列。本发明所使用的术语“非编码元件”也可以被称为“非编码序列”,是指细胞基因组中存在的不编码氨基酸的核苷酸序列,它可以是非编码的基因,也可以是非基因的其它非编码元件,包括但不限于lncRNA基因、microRNAs、顺式调控元件和其它未注释的元件。本发明所使用的术语“lncRNA基因”也可以简写为“lncRNA”,是指长链非编码RNA(longnon-codingRNA),是长度大于200个核苷酸的非编码RNA。术语“非编码RNA”是指不能翻译为蛋白的功能性RNA分子,包括但不限于小干涉RNA、长链非编码RNA等。研究表明,lncRNA在剂量补偿效应(Dosagecompensationeffect)、表观遗传调控、细胞周期调控和细胞分化调控等众多生命活动中发挥重要作用,是遗传学研究的热点。本文使用的术语“功能性lncRNA基因”是指在细胞中发挥作用、具有功能的lncRNA基因。本文使用的术语“成对基因”或“功能性成对基因”是指一对基因,只有当两个基因都被敲除时才能表现出与功能相关的表型,而仅敲除一个基因无法表现出与功能相关的表型。成对基因可以是具有相同的或类似的功能的基因,当仅敲除一个基因时,由于另一个基因的补偿作用,导致无法表现出与功能相关的表型。在一些实施方案中,所述成对基因是成对编码基因。Cas9/CRISPR系统利用RNA指导的DNA结合对靶DNA进行序列特异性切割,由crRNA(CRISPR-derivedRNA)通过碱基配对与tracrRNA(trans-activatingRNA)结合形成tracrRNA/crRNA复合物,此复合物引导核酸酶Cas9蛋白在与crRNA配对的靶序列上的特定位置处剪切双链DNA。与crRNA配对的靶序列通常是位于基因组PAM(原间隔区邻近基序)位点(NNG)上游的约20个核苷酸的序列。Cas9蛋白对靶位点的切割需要借助向导RNA。术语“向导RNA”又称gRNA(guideRNA),gRNA通常包括crRNA上与靶序列互补的核苷酸和由crRNA与tracrRNA碱基配对形成的RNA骨架(Scaffold),能够识别与crRNA配对的靶序列。gRNA可以与Cas9蛋白形成复合体并将Cas9蛋白带至靶序列并切割其中的靶位点。传统上,gRNA通常是sgRNA(singleguideRNA)的形式。sgRNA又称“单链向导RNA”,是crRNA和trancrRNA融合而成的一条RNA链。通常,如本领域技术人员所熟知的,sgRNA包含与靶序列配对的序列(也被称为gRNA配对序列或sgRNA配对序列)、骨架(Scaffold)序列(也被称为gRNA骨架序列)和转录终止子(例如polyT)。本发明中,除非特别指出,gRNA和sgRNA可以互换使用。本文所使用的术语“gRNA骨架序列”或“骨架序列”是指sgRNA中gRNA配对序列与转录终止子之间的序列。gRNA(sgRNA)的设计方法和设计工具是本领域众所周知的,可以通过多种途径获得,例如但不限于HorizonDiscovery公司提供的gUIDEbood在线平台。本发明所使用的术语“pgRNA”是指成对gRNA(pairedRNA)或者成对sgRNA,是指一对gRNA(或者sgRNA)。在一些实施方案中,本发明中所述的pgRNA是一对靶向同一个核酸片段上的不同靶位点或者分别靶向不同核酸片段的gRNA(或者sgRNA)。在一些实施方案中,本发明中所述的pgRNA是一对靶向同一个非编码元件上的不同靶位点的gRNA(或者sgRNA)。优选地,一个pgRNA对中两个gRNA的靶位点之间的间隔为200bp-10kb。在另一些实施方案中,本发明中所述的pgRNA是一对分别靶向成对基因中的两个基因的gRNA(或者sgRNA)。本发明可以应用Cas9/CRISPR系统在pgRNA所针对的两个靶位点上同时进行切割,达到基因敲除的目的。例如,本发明可以应用Cas9/CRISPR系统在pgRNA所针对的两个靶位点之间的片段进行切割,从而实现对这两个靶位点之间的序列的删除,达到基因敲除的目的(例如靶位点在非编码元件上的情况)。再例如,本发明还可以应用Cas9/CRISPR系统对pgRNA所针对的两个靶基因分别进行切割,从而实现对这两个靶基因的敲除(例如两个靶基因是成对基因的情况)。应用本发明所构建的pgRNA表达质粒文库可以实现对多个核酸序列,例如非编码元件(如lncRNA基因)或者多对成对基因进行高通量的敲除,从而实现对功能性核酸序列,例如功能性非编码元件或功能性成对基因的高通量筛选。在一些实施方案中,本发明提供构建pgRNA表达质粒文库的方法,所述文库中包含的每个pgRNA表达质粒上具有顺序连接的分别与各自的U6启动子可操作连接的两个gRNA序列,这两个gRNA组成一个pgRNA,靶向一个核酸序列或靶向不同的核酸序列,例如靶向一个非编码元件(例如一个lncRNA基因)或靶向成对基因(其中两个gRNA分别靶向成对基因中的两个基因),。本发明的文库构建方法是通过两步连接反应实现的,首先提供包含顺序连接的第一U6启动子、第二gRNA骨架序列编码序列和转录终止子的质粒(即初始质粒),然后通过第一步连接反应将多个“第一gRNA配对序列编码序列-间隔序列-第二gRNA配对序列编码序列”的每一个分别插入到该质粒上的第一U6启动子和第二gRNA骨架序列编码序列之间,然后转化感受态细胞获得第二质粒混合物,然后再通过第二步连接反应将顺序连接的第一gRNA骨架序列、转录终止子和第二U6启动子插入到第二质粒中两个gRNA配对序列之间,再转化感受态细胞获得pgRNA表达质粒文库,其中每个pgRNA表达质粒包含顺序连接的“第一U6启动子-第一gRNA配对序列-第一gRNA骨架序列-转录终止子-第二U6启动子-第二gRNA配对序列-第二gRNA骨架序列-转录终止子”的序列。本发明中第一U6启动子和第二U6启动子可以是相同的或不同的。本发明中第一gRNA骨架序列和第二gRNA骨架序列可以是相同的或不同的。本发明所使用的术语“质粒”也可以被称为表达载体,当其被转入细胞中时,可以表达其中所包含的与启动子相连的编码序列。质粒中通常含有基因表达所必须的元件。本发明中,所使用的质粒可以是慢病毒载体质粒,或者任何其它可以表达其中所含编码基因的质粒,优选为慢病毒载体质粒。本发明所使用的术语RNA(例如pgRNA或gRNA骨架序列或gRNA配对序列)的“编码序列”是指编码该RNA的DNA序列。本发明使用的术语“转录终止子”是指给予RNA聚合酶转录终止信号的DNA序列。可用的转录终止子是本领域技术人员众所周知的。本发明可以使用任何合适的转录终止子。本发明所使用的转录终止子优选为polyT。在一些实施方案中,第一gRNA配对序列或第二gRNA配对序列的靶位点独立地位于靶基因的启动子或基因本体上。在一些实施方案中,所述pgRNA靶向非编码元件。在一些实施方案中,所述pgRNA表达质粒文库靶向超过50个非编码元件,优选超过100个非编码元件、超过200个非编码元件,超过500个非编码元件,甚至超过1000个非编码元件。在一些实施方案中,所述pgRNA表达质粒文库中不同的pgRNA靶向不同的非编码元件,或者多个pgRNA靶向同一个非编码元件,或者是二者的组合。在一些实施方案中,所述非编码元件是非编码基因,优选lncRNA基因。在一些实施方案中,所述pgRNA靶向成对基因。即,pgRNA中所包含的两个gRNA分别靶向成对基因中的两个基因。在一些实施方案中,所述pgRNA表达质粒文库靶向超过50对成对基因,优选超过100对成对基因、超过200对成对基因,超过500对成对基因,甚至超过1000对成对基因,或者甚至超过10000对成对基因。在一些实施方案中,所述成对基因优选是成对编码基因。在一些实施方案中,初始质粒中的第一U6启动子和第二gRNA骨架序列编码序列之间可以含有在其两端具有限制性核酸内切酶酶切位点的自杀基因。所述限制性核酸内切酶优选为II型限制性核酸内切酶,更优选为BsmBI。本发明所使用的术语“自杀基因”是指在宿主细胞中表达时对宿主细胞具有致死作用的基因。自杀基因可以被构建到表达载体上,作为转化时的选择标记,自杀基因的表达产物能够抑制宿主细胞生长,在克隆是没有被切开或自身环化的质粒转化的宿主细胞不能生长。优选使用的自杀基因是ccdB基因,一种毒素蛋白基因,例如Invitrogen公司推出的载体转化系统。在一些实施方案中,所述第一步连接反应是将在第一U6启动子的3'端和第二gRNA骨架序列编码序列的5'端被切割的初始质粒与多个DNA寡核苷酸序列的混合物进行连接反应。在优选的实施方案中,初始质粒被限制性核酸内切酶切割。初始质粒首先被限制性核酸内切酶,优选II型(TypeIIs)限制性核酸内切酶,更优选BsmBI切割,以使得初始质粒在第一U6启动子的3'端和第二gRNA骨架序列编码序列的5'端被切割,然后再与多个DNA寡核苷酸序列的混合物进行连接反应。所述多个DNA寡核苷酸序列的每一个包含一个pgRNA的两个gRNA配对序列,即第一gRNA配对序列、第二gRNA配对序列的编码序列,并且包含顺序连接的“第一gRNA配对序列编码序列-间隔序列-第二gRNA配对序列编码序列”的序列。所述DNA寡核苷酸序列的间隔序列中包含切割位点(例如限制性核酸内切酶切割位点,优选II型(TypeIIs)限制性核酸内切酶切割位点,更优选BsmBI切割位点),以便于在第一gRNA配对序列编码序列和第二gRNA配对序列编码序列之间进行切割以进行第二步连接反应。所述DNA寡核苷酸序列的两端还可以具有与引物配对的序列,以便于通过引物配对介导的扩增反应扩增所述DNA寡核苷酸序列。在进行第一步连接反应之前,优选对所述DNA寡核苷酸序列混合物进行扩增。在一些实施方案中,第一步连接反应通过Gibson组装方法进行。此时,扩增所述DNA寡核苷酸序列混合物所使用的引物两端具有进行Gibson组装所需的与经过限制性核酸内切酶切割的初始质粒同源的序列。本发明中所使用的术语“Gibson组装”又称为Gibson由J.CraigVenter研究所的DanielGibson博士在2009年提出。Gibson组装方法适合于拼接多个线性DNA片段,也适合将目的DNA插入载体中。进行Gibson组装时,首先需要在DNA片段的末端加上同源片段,然后将这些DNA片段和mastermix混合孵育一个小时,mastermix由三种酶组成,其中DNA外切酶先从5'端降解核苷酸产生粘性末端,然后相邻片段之间的重叠序列退火,最后DNA聚合酶和DNA连接酶将序列填充,形成完整的双链DNA分子,实现无痕拼接。mastermix可以从NEB或者SGI-DNA公司购买,或者自己配制(参见MillerLabProtocol)。在一些实施方案中,第一步连接反应的反应混合物转化感受态细胞获得第二质粒混合物。第二质粒混合物中包含的每一个第二质粒包含顺序连接的“第一U6启动子-第一gRNA配对序列编码序列-间隔序列-第二gRNA配对序列编码序列-第二gRNA骨架序列编码序列-转录终止子”序列。在一些实施方案中,每一个第二质粒中在第一U6启动子和第一gRNA配对序列编码序列之间,以及第二gRNA配对序列编码序列和第二gRNA骨架序列编码序列之间是紧邻连接。在一些实施方案中,所述第二步连接反应是将在第一gRNA配对序列编码序列的3'端和第二gRNA配对序列编码序列的5'端被切割的第二质粒混合物与包含“第一gRNA骨架序列编码序列-转录终止子-第二U6启动子”序列的DNA片段进行连接反应。在优选的实施方案中,第二质粒被限制性核酸内切酶,优选II型(TypeIIs)限制性核酸内切酶,更优选BsmBI切割。第二质粒混合物首先被限制性核酸内切酶,优选II型(TypeIIs)限制性核酸内切酶,更优选BsmBI切割,以使得第二质粒在第一gRNA配对序列编码序列的3'端和第二gRNA配对序列编码序列的5'端被切割,然后再与包含“第一gRNA骨架序列编码序列-转录终止子-第二U6启动子”序列的DNA片段进行连接反应。所述包含“第一gRNA骨架序列编码序列-转录终止子-第二U6启动子”序列的DNA片段中优选在转录终止子和第二U6启动子之间还包含接头序列,所述接头序列可以包含引物结合区域,用于扩增含有第一gRNA配对序列编码序列和/或第二gRNA配对序列编码序列的片段。在一些实施方案中,第二步连接反应得到的反应混合物转化感受态细胞获得pgRNA表达质粒文库。pgRNA表达质粒文库中的每个pgRNA表达质粒包含顺序连接的“第一U6启动子-第一gRNA配对序列编码序列-第一gRNA骨架序列编码序列-转录终止子-第二U6启动子-第二gRNA配对序列编码序列-第二gRNA骨架序列编码序列-转录终止子”的序列。其中“第一gRNA配对序列-第一gRNA骨架序列-转录终止子”组成第一gRNA(或sgRNA)序列,“第二gRNA配对序列-第二gRNA骨架序列-转录终止子”组成第二gRNA(或sgRNA)序列。在一些实施方案中,每一个pgRNA表达质粒中第一U6启动子和第一gRNA配对序列编码序列之间,以及第一gRNA配对序列和第一gRNA骨架序列编码序列之间是紧邻连接。在一些实施方案中,每一个pgRNA表达质粒中第二U6启动子和第二gRNA配对序列编码序列之间,以及第二gRNA配对序列和第二gRNA骨架序列编码序列之间是紧邻连接。本发明中,“第一序列-第二序列-第三序列”以及与其类似的表述方式表示其所包含的各个序列按照文字所述的顺序连接。在一些实施方案中,各个序列之间可以具有或不具有其它核苷酸或核苷酸序列。优选地,在本发明中,在文库构建方法中所使用的和所产生的质粒和DNA片段中,以及在最终制备的pgRNA表达质粒文库中,相邻连接的gRNA配对序列编码序列和gRNA骨架序列编码序列之间,以及gRNA配对序列编码序列和转录启动子之间均是紧邻连接。本发明所使用的术语“紧邻连接”或“紧邻”是指顺序相连的两个元件(例如DNA序列)之间不存在其它任何核苷酸。本发明所使用的术语“顺序连接”是指两个或更多个元件(例如DNA序列)按照文字所示的顺序连接。在一些实施方案中,顺序连接的两个或更多个元件(例如DNA序列)可以具有或不具有其它核苷酸或核苷酸序列。本发明所使用的初始质粒还可以包含与启动子可操作相连的标记物基因,以用于筛选含有pgRNA表达质粒的细胞。本发明所使用的术语“标记物基因”是指其表达可以被选择或富集的任何标记物基因,即当该标记物基因在细胞中表达时,可以通过一定方式选择和富集表达该标记物基因的细胞。可用于本发明的标记物基因包括但不限于在表达后可以用FACS分选的荧光蛋白基因,或者可以利用抗生素进行筛选的抗性基因,或者表达后可以被对应抗体识别并通过免疫染色或磁珠吸附进行筛选的蛋白基因。可用于本发明的抗性基因包括但不限于针对杀稻瘟菌素(Blasticidin)、遗传霉素(Geneticin,G-418)、潮霉素(HygromycinB)、霉酚酸(MycophenolicAcid)、嘌呤霉素(Puromycin)、博莱霉素(Zeocin)或新霉素(Neomycin)的抗性基因。可用于本发明的荧光蛋白基因包括但不限于蓝色荧光蛋白(CyanFluorescentProtein)、绿色荧光蛋白(GreenFluorescentProtein)、增强绿色荧光蛋白(EnhancedGreenFluorescentProtein)、黄色荧光蛋白(YellowFluorescentProtein)、橙色荧光蛋白(OrangeFluorescentProtein)、红色荧光蛋白(RedFluorescentProtein)、远红色荧光蛋白(Far-RedFluorescentProtein)或可切换荧光蛋白(SwitchableFluorescentProteins)的基因,优选为增强绿色荧光蛋白(EGFP)。与标记物基因可操作连接的启动子可以是本领域中经常用于标志物基因表达的任何启动子,包括但不限于CMV启动子。在一个优选的实施方案中,初始质粒是慢病毒载体,包含从5'到3'依次为第一U6启动子,两端具有BsmBI酶切位点的ccdB基因、第二gRNA骨架序列编码序列、polyT、CMV启动子、EGFP基因。DNA寡核苷酸序列中含有一对pgRNA的两个gRNA配对序列编码序列,两个gRNA配对序列编码序列之间的间隔序列两端具有BsmBI酶切位点。DNA寡核苷酸序列的两端具有引物配对序列。包含“第一gRNA骨架序列编码序列-转录终止子-第二U6启动子”序列的DNA片段中在第一gRNA骨架序列编码序列的5'端和第二U6启动子的3'端具有BsmBI酶切位点,并且在转录终止子和第二U6启动子之间具有接头序列。用靶向DNA寡核苷酸序列侧翼序列的引物进行扩增,其中两个引物上分别具有与第一U6启动子3'区域和第二gRNA骨架序列编码序列5'区域同源的序列,以产生与BsmBI消化的初始质粒同源的60bp同源序列。扩增的DNA寡核苷酸序列混合物通过Gibson组装方法被连接到初始质粒中,并被转化到Trans1-T1感受态细胞中以获得第二质粒,然后用BsmBI消化第二质粒,并与用BsmBI消化的包含“第一gRNA骨架序列编码序列-转录终止子-第二U6启动子”序列的DNA片段相连,将连接混合物转化到Trans1-T1感受态细胞中,以获得最终的文库质粒。利用上述方法构建获得的pgRNA表达质粒文库也在本发明的范围内。在一些实施方案中,本发明提供构建核酸序列,例如非编码元件(例如lncRNA基因)或成对基因敲除细胞文库的方法,包括,将本发明的pgRNA表达质粒文库递送到靶细胞中,并向靶细胞引入Cas9核酸酶,培养细胞后筛选成功转入所述pgRNA表达质粒文库的细胞,获得核酸序列,例如非编码元件(例如lncRNA基因)或成对基因敲除细胞文库。将质粒文库递送到靶细胞中的方法是本领域众所周知的,例如但不限于通过电穿孔、显微注射、基因枪、磷酸钙共沉淀、脂质体转染、病毒介导的转染技术。病毒介导的转染技术可以通过逆转录病毒、腺病毒或慢病毒转染。慢病毒转染需要使用慢病毒载体制备质粒文库,然后将质粒文库与慢病毒包装质粒共转染宿主细胞,产生质粒文库的慢病毒,然后再用慢病毒转导靶细胞,感染后筛选侵染成功的靶细胞。所使用的宿主细胞可以是任何可用于被慢病毒在体细胞和慢病毒包装质粒共转染以产生慢病毒的细胞,包括但不限于HEK293T细胞。在本发明的一些实施方案中,通过慢病毒感染将所述pgRNA表达质粒文库递送到靶细胞中。在一些实施方案中,将所述pgRNA表达质粒文库与病毒包装质粒共转染宿主细胞,产生含有所述pgRNA表达质粒文库的慢病毒,用该慢病毒感染靶细胞,从而使所述pgRNA表达质粒文库递送到靶细胞中。在一些实施方案中,慢病毒感染靶细胞的MOI小于等于0.3,以使得一个慢病毒感染一个靶细胞。在一些实施方案中,感染细胞到收集被感染细胞之间的培养时间为约48h至约72h。在一些实施方案中,通过药物抗性或FACS方法收获受感染的细胞。通过上述方法构建获得的核酸序列敲除细胞文库,例如非编码元件或成对基因敲除细胞文库也在本发明的范围之内。本发明中,Cas9核酸酶可以以蛋白质的形式或者以其编码核酸序列(例如mRNA或cDNA)的形式被引入细胞。编码Cas9的核酸可以被包含在质粒或病毒载体(例如慢病毒载体)中被引入细胞,例如通过转染被引入细胞。编码Cas9的核酸也可以通过电穿孔、脂质体、显微注射等方式被直接递送到细胞中。本发明中,可以将Cas9和pgRNA表达质粒文库同时引入细胞,或者,例如,可以先将Cas9引入细胞,再将pgRNA表达质粒文库引入细胞。在一些实施方案中,用包含Cas9的载体和pgRNA表达质粒文库共转染细胞。在另一些实施方案中,将Cas9和pgRNA表达质粒文库在体外组装成复合体,然后转染细胞。在其它的实施方案中,先将Cas9在细胞中稳定表达,再用pgRNA表达质粒文库转染细胞。本发明所述的细胞可以是任何真核细胞,例如分离的动物细胞,例如全能细胞、多能细胞、成体干细胞、受精卵或体细胞等。在一些实施方案中,所述细胞是脊椎动物细胞。在一些实施方案中,所述细胞是哺乳动物细胞。在一些实施方案中,所述细胞是人细胞,例如HEK293T细胞、HeLa细胞、Huh7.5OC细胞等。在一些实施方案中,所述细胞是牛、山羊、绵羊、猫、狗、马、啮齿类动物、鱼、灵长类动物的细胞。在一些实施方案中,啮齿类动物包括小鼠、大鼠、兔。在一些实施方案中,本发明还提供筛选功能性核酸序列,例如功能性非编码元件(例如lncRNA基因)或功能性成对基因的方法,包括:培养上述细胞文库或在特定的筛选条件下培养上述细胞文库,然后提取细胞文库混合物的基因组DNA,扩增包含pgRNA中的任何一个或两个gRNA配对序列编码序列的DNA片段,利用深度测序技术对扩增产物进行测序,分析测序结果,从而确定pgRNA的靶核酸序列,例如靶非编码元件(例如lncRNA基因)或靶成对基因的功能。在一些实施方案中,所述培养的时间足以使所希望筛选的核酸序列的功能被检测到。在一些实施方案中,扩增包含pgRNA中的任何一个或两个gRNA配对序列编码序列的DNA片段的目的是为了对这些DNA片段进行深度测序,从而鉴别培养的细胞文库中存在包含哪些pgRNA的细胞及其比例。可以以pgRNA表达质粒中第一U6启动子之前的序列、转录终止子和第二U6启动子之间的接头序列、和/或第二gRNA骨架序列编码序列之后的序列为引物的靶序列,进行扩增。本文所使用的术语“深度测序”又可被称为“高通量测序”或“下一代测序”,可以一次对几十万到几百万条DNA分子平行地进行序列测定。深度测序的测序结果可以是各个不同测序DNA片段的reads数。深度测序技术是本领域中的成熟技术,有多个厂商提供深度测序服务,或者提供用于进行深度测序的试剂盒、仪器和说明书。可以使用的深度测序技术包括但不限于在IonTorrent或Illumina的测序平台上进行的深度测序,例如使用IlluminaHiSeq2500进行深度测序。通过深度测序可以对扩增获得的细胞文库中包含的所有pgRNA相关序列(即包含pgRNA中的任何一个或两个gRNA配对序列编码序列的DNA片段)进行测序。深度测序的测序结果可以是各个不同测序DNA片段的reads数或各个不同测序DNA片段的占比。各个不同测序DNA片段(例如各个不同pgRNA相关序列)的“占比”可以指各个不同测序DNA片段(例如各个不同pgRNA相关序列)的reads数与所有测序DNA片段(即扩增获得的所有pgRNA相关序列)的reads数之比。根据深度测序结果,如果某些pgRNA与对照相比占比提高,则说明包含这些pgRNA的细胞在培养过程中被富集,其靶核酸序列的敲除有利于细胞增殖;如果某些pgRNA与对照相比占比降低,则说明包含这些pgRNA的细胞在培养过程中逐渐减少,其靶核酸序列的敲除不利于细胞增殖。本发明的细胞文库可用于筛选功能性核酸序列,例如非编码元件(例如lncRNA基因)或成对基因,例如用于筛选调控细胞增殖的功能性核酸序列,例如非编码元件(例如lncRNA基因)或成对。所述功能包括但不限于,例如,该核酸序列的敲除促进细胞生长或抑制细胞生长,或者该核酸序列的敲除导致细胞生长被特定药物抑制或被特定药物促进。所述“特定的筛选条件”包括但不限于在存在某种药物的条件下,或者在存在某种蛋白质的条件下等。在一种实施方案中,例如,培养本发明的核酸序列敲除细胞文库,然后提取细胞文库混合物的基因组DNA,扩增包含pgRNA对中的任何一个或两个gRNA配对序列编码序列的DNA片段,利用深度测序技术对扩增产物进行测序,分析测序结果,以培养起始时的细胞文库测序结果作为对照,测序结果中与对照相比占比提高的pgRNA说明其靶核酸序列的敲除有利于细胞增殖,测序结果中与对照相比占比降低的pgRNA说明其靶核酸序列的敲除不利于细胞增殖。在另一种实施方案中,例如,在特定的筛选条件下培养本发明的核酸序列敲除细胞文库,然后提取细胞文库混合物的基因组DNA,扩增包含pgRNA对中的任何一个或两个gRNA配对序列编码序列的DNA片段,利用深度测序技术对扩增产物进行测序,分析测序结果,以不在该特定筛选条件下培养的细胞文库测序结果为对照,测序结果中与对照相比占比提高的pgRNA说明其靶核酸序列的敲除有利于细胞在该筛选条件下增殖,测序结果中与对照相比占比降低的pgRNA说明其靶核酸序列的敲除不利于细胞在该筛选条件下增殖。在另一种具体的实施方案中,例如,在存在特定药物的条件下培养本发明的核酸序列敲除细胞文库,然后提取细胞文库混合物的基因组DNA,扩增包含pgRNA对中的任何一个或两个gRNA配对序列编码序列的DNA片段,利用深度测序技术对扩增产物进行测序,分析测序结果,以不在该特定筛选条件下培养的细胞文库为对照,测序结果中与对照相比占比提高的pgRNA说明其靶核酸序列的敲除有利于细胞在该筛选条件下增殖,即其靶核酸序列的敲除可提高细胞对该特定药物的耐受性;测序结果中与对照相比占比降低的pgRNA说明其靶核酸序列的敲除不利于细胞在该筛选条件下增殖,即其靶核酸序列的敲除可降低细胞对该特定药物的耐受性。就测序结果的比较而言,术语“对照”是指能反映出所研究的核酸序列的功能的对照,这种对照的选择属于本领域技术人员的常规技术。例如当检测核酸序列的敲除对于细胞增殖的影响时,以培养起始时的细胞文库测序结果作为对照。再例如当检测核酸序列的敲除对于细胞在特定筛选条件下的增殖的影响时,以不在该特定筛选条件下培养的细胞文库测序结果为对照。本发明(例如实施例)中所使用的术语“正向筛选”是指靶核酸序列,例如非编码元件(例如lncRNA基因)的敲除导致促进细胞生长。本发明(例如实施例)中所使用的术语“负向筛选”是指靶核酸序列,例如非编码元件(例如lncRNA基因)的敲除导致抑制细胞生长。本发明中所使用的术语“包括”或“包含”表示“包括但不限于”、“基本上由……组成”或“由……组成”。结合以下实施例和附图对本发明进行进一步说明,它们仅用于举例说明,并非要限制本发明的范围。如果没有特别指明,实施例均按照常规实验条件,如Sambrook等分子克隆实验手册(SambrookJ&RussellDW,Molecularcloning:alaboratorymanual,2001),或按制造商提供的说明进行。实施例1通过慢病毒递送成对向导RNA(pairedgRNA,pgRNA)1.构建CRISPRpgRNA文库,以使得两个gRNA靶位点之间的基因组序列被同时删除测试了两种方法以在一个慢病毒骨架中表达pgRNA,一种是用两个U6启动子分别驱动两个gRNA(U62),另一种是用单个U6启动子驱动两个顺序相连的gRNA(U61)(图1a)。为了对这两种方法进行比较,使用六对靶向CSPG4基因的gRNA(参见下列表1和表2,图1b)诱导大片段删除,预测它们将会删除人CSPG4基因座的2kb-4.5kb(基因CSPG4编码完整膜硫酸软骨素蛋白聚糖)(图1b和c)。表1靶向CSPG4的用于DNA片段删除的成对gRNA(pgRNA)的设计表2靶向CSPG4的用于DNA片段删除的成对gRNA(pgRNA)的序列sgRNACSPG4序列sgRNA15’-AGGAGACTGGAGGTAAGACAsgRNA1’5’-TCACTCCTGTGCACAGCAGCsgRNA25’-AGAAGAGCTGGCCCAGCAGCsgRNA2'5’-CCACCACATACACACCTATGsgRNA35’-AGTCTAGTGAGACGGAGGCGsgRNA3’5’-TGCTGGGAGGAGGTTTGAGAsgRNA45’-TCAGTCTCGGGATCTCTGATsgRNA4’5’-TGGCCAGTGATGAGCCTTCTsgRNA55’-GTGCTGGGACTTGCTGTGGTsgRNA5’5’-CAGAAAGGCAACTAAACAGAsgRNA65’-ACACCTCTTGCCAGTCTGCTsgRNA6’5’-GTTGTAAGCTCCATGGGATTsgRNAAAVS15’-CGGAACCTGAAGGAGGCGGC在稳定表达Cas9和OCT1基因(OCT1基因编码转录因子)的肝癌细胞系Huh7.5OC[8,16]中,U62载体中的全部六个pgRNA都产生了正确大小的基因组删除,但是U61载体中只有两个pgRNA产生正确的删除,其效率低得多(图1c),这表示使用U62更佳,因此使用U62进行后续的实验。发现U62中靶向lncRNAMALAT1的五个pgRNA(参见表3和表4)也以高效率产生正确大小的基因组删除(图2a和b)。表3靶向MALAT1的用于DNA片段删除的成对gRNA(pgRNA)的设计表4靶向MALAT1的用于DNA片段删除的成对gRNA(pgRNA)的序列sgRNAMALAT1序列sgRNA15’-CCGCAGATCAGAGTGGGCCACsgRNA1’5’-GGATAGTACACTTCACTCAGsgRNA25’-ACACAAGAAGTGCTTTAAGsgRNA2'5’-GGGATCAAGTGGATTGAGGsgRNA35’-CCCGAATTAATACCAATAGAsgRNA3’5’-CTTGAATGTCTCTTAGAGGGsgRNA45’-CCCATCAATTTAATTTCTGGsgRNA4’5’-CCAGTTTGAATTGGGAAGCTsgRNA55’-GAGCCAGTGCGATTTGGTGAsgRNA5’5’-GGTCTTAACAGGGAAGAGAG接下来研究了慢病毒递送的pgRNA的转导后培养时间是否影响基因删除的效率,并观察到随时间持续进行的基因组删除,并在转导后大约15天达到平台期(图1d)。当用另一个靶向CSPG4的pgRNA(2+2’,图1b)和另一个靶向MALAT1的pgRNA(2+2’,图2a,c和d)诱导基因组删除时,获得了类似的结果。因此,转导后培养文库细胞至少2周是合乎需要的,使得有足够的时间在哺乳动物细胞中产生基因组删除,删除的水平适合于筛选。对五个pgRNA靶向区域(其中3个pgRNA靶向CSPG4,2个pgRNA靶向MALAT1)进行总基因组测序,发现每一个位点上几乎有80%的删除是在前间区序列邻近基序(PAM)上游3nt的两个Cas9切割位点的准确连接处(图1e和图2e),这与之前的发现一致[17]。这些结果一起表明,慢病毒递送的pgRNA能够在哺乳动物中高效产生大片段的基因组删除。实施例2pgRNA文库构建和基因组范围内的lncRNA删除筛选设计了一种pgRNA文库,其靶向大约700个lncRNA基因(表5),这些基因在癌症或其它疾病中具有已知的或推测的作用[18]。表5用于功能筛选的pgRNA文库的总结我们开发了一种快速和精确的方法,来将pgRNA克隆到慢病毒表达载体中(图3a,图4a和4b)。由于每一个gRNA对中的两个gRNA由同一种类型的U6启动子驱动,并含有相同的3’骨架序列,可能发生重组,这会导致错误的pgRNA装配。因此测试了两种pgRNA文库质粒构建体中的重组率和转导后细胞中染色体的整合,发现细胞中病毒转导后发生了重组,重组率为大约7.5%,这与寡核苷酸(oligo)合成错误率相当(表6)。这表明重组对于pgRNA文库筛选的影响可以忽略不计。表6pgRNA文库的定性总结类别质粒文库细胞文库成功率92.5%84.2%突变率7.5%8.3%重组率0%7.5%注:对80个和120个分别从质粒和细胞文库中随机选取的gRNA的序列进行验证。在U62中构建pgRNA文库,将其以低的感染复数(MOI=0.3)(感染复数较低以使得一个细胞仅转入一对gRNA)转入到Huh7.5OC中,Huh7.5OC之前曾被用于对编码基因进行功能性筛选[16]。转导后培养30天以试图鉴别对细胞的生长和生存力具有积极影响或消极影响的lncRNA并使这些lncRNA的鉴别达到最大的程度。在CRISPR筛选之前或之后从提取的细胞基因组DNA中对gRNA编码区域进行PCR扩增,以进行深度测序分析(图3b和图4c)。总体来说,每一种条件下的3个独立实验重复的read分布显示高水平的相关性(图6)。30天的培养之后,与阴性对照pgRNA(非靶向pgRNA或靶向非功能性AAVS1基因座的pgRNA)相比,靶向阳性对照基因(大部分是核糖体基因)或lncRNA的pgRNA被耗尽(即转入该pgRNA的细胞减少)(图5a和图5b),这表明了它们对于细胞存活或增殖的效果。使用MAGeCK算法通过比较第30天的样品和第0天的对照来鉴别排名靠前的被选中的lncRNA基因(thetophits)[21]。MAGeCK使用负二项式(negativebinomial,NB)模型来评估单个pgRNA丰度变化的统计学显著性,并使用均匀分布的零模型比较靶向每一个lncRNA的pgRNA的排名(ranks)(参见下文“材料与方法”部分)。MAGeCK的输出是一组阴性(或阳性)选择的lncRNA,或者其敲除破坏(或刺激)细胞增殖的lncRNA。MAGeCK鉴别了43个负向筛选的和8个正向筛选的具有统计学显著性的lncRNA(错误发现率<0.25)。基因集合富集分析(GeneSetEnrichmentAnalysis,GSEA)表明在负向筛选pgRNA的排序列表中阳性对照pgRNA显著富集,如所期望的那样显示了它们的靶基因的基本功能[22]。排名靠前的负向筛选基因包括两个阳性对照基因:RPL18A,一个必须基因,和EZH2,一个编码多梳家族成员的基因,它在肝癌细胞的增殖中具有重要作用[23]。靶向RPL18A和EZH2的启动子和外显子的pgRNA一起被耗尽(图5b)。类似地,当76%的靶向正向筛选的lncRNA被富集的时候,89%的靶向排名靠前的负向筛选的lncRNA的pgRNA被耗尽(图5c和5d,图7a和7b)。相反,非靶向对照的pgRNA和靶向AAVS1基因座的pgRNA的丰度在对照和处理条件下是类似的(图7c,表7)。表7阴性对照pgRNA的筛选结果有趣的是,靶向必需基因的内含子区域的266个pgRNA降低了细胞生存力(图5b),这可能是由于调控元件的删除或靶基因的可变剪接的调控[24,25]。实施例3被选择的lncRNA候选者的验证从正向或负向筛选的具有统计学显著性的lncRNA中,获得了排名靠前的被选中的lncRNA基因(hits),在3个独立的实验重复中,它们相应的pgRNA分别被一致地耗尽(对于负向筛选)或富集(对于正向筛选)(图5c和5d和图8,表8和表9)。表8负向筛选的lncRNA基因表9正向筛选的lncRNA基因为了验证这些lncRNA中的一些的功能,选了筛选文库中原始存在的两对gRNA,并为每个基因设计了多达3种其它的新的pgRNA。此外,设计3对gRNA靶向AAVS1基因座,作为阴性对照(表10)。表10被选择的lncRNA的功能验证的pgRNA设计用携带CMV-EGFP的慢病毒骨架将所有pgRNA重新转导到Huh7.5OC细胞中。根据EGFP阳性细胞的百分比变化对细胞的增殖进行定量。来自负向筛选列表中排名第一的核糖体基因,RPL18A的启动子的删除,显著减少了细胞增殖,AAVS1基因座的删除对细胞生长的作用可以忽略不计(图9a)。使用相同的方法,从pgRNA文库筛选中选择与编码基因没有任何重叠的lncRNA进行验证。选择了5个lncRNA,在初步的筛选中,它们的删除似乎抑制了细胞的增殖:AC004463.6、AC095067.1、HM13-AS1、RP11-128M1.1和RP11-439K3.1。还选择了4个lncRNA,它们的删除似乎正向调节细胞生长:LINC00176、LINC01087、LINC00882和LINC00883。设计pgRNA来靶向lncRNA的启动子或外显子。对于分开转录但共用同一个启动子的一对lncRNA:LINC00882andLINC00883,设计另外3个pgRNA靶向外显子。根据单个删除的结果,发现来自负向筛选筛选的所有5个lncRNA对于细胞增殖是必不可少的,来自正向筛选筛选的所有4个lncRNA被证明负调节细胞增殖(图9b和9c,图10a和10b)。将LINC00882的cDNA克隆引入到两组包含靶向LINC00882的pgRNA并且删除了LINC00882的Huh7.5OC细胞中,并证明LINC00882的外源回补表达可以负调节细胞增殖(图10c和10d)。一些pgRNA不产生表型,例如RP11-439K3.1_p3和RP11-439K3.1_p4(图9b),而且已证明这是因为这些pgRNA不能产生基因组删除(图10e)。为了进一步对候选基因进行验证,使用CRISPR抑制剂(CRISPRi)方法[26],这种方法能够减少靶区域的转录。发现使用CRISPRi对三个负向筛选的lncRNA(AC004463.6、RP11-439K3.1和AC095067.1)的阻抑显著降低了细胞增殖(图9d)。还对五个敲除了负向筛选的lncRNA的细胞系和CRISPRi细胞系(lncRNA的转录被阻抑)进行了细胞死亡信号检测,发现所有5个lncRNA对于细胞存活来说是必需的(图11和表11)。表11用于对选择的lncRNA进行功能验证的CRISPR抑制和CRISPR上调的sgRNA设计对于正向筛选的基因候选者,我们使用CRISPR上调(CRISPRa)方法[27]对LINC01087和LINC00882的转录进行正调节,发现这两个lncRNA的过表达都是致命的(图12和表11)。因此,基因组删除的CRISPR/Cas9筛选策略对于负向和正向筛选的lncRNA都是有效的,具有高效性和可靠性。对于文库筛选和候选者验证,将成对gRNA引入细胞。有可能观察到的表型变化是由于一个gRNA介导的双链断裂(DSB)的效果,而不是pgRNA介导的基因组删除的效果。为了排除这种可能性,我们比较了靶向AC004463.6和AC095067.1的pgRNA的效果和引入它们相应的gRNA中的仅仅一个的效果。在两种情况下,只有pgRNA显著影响细胞增殖,而靶向内含子或外显子的单个gRNA没有改变细胞生存(图9e和图10f)。这表明至少对于这两个基因座,pgRNA-介导的基因组删除对于产生lncRNA的功能性敲除是必需的,由单个gRNA产生的indels不太可能获得这样的效果。使用这种方法,筛选到了大约700个人lncRNA,并可以鉴别在癌细胞中具有致癌作用或肿瘤抑制作用的lncRNA。应用该筛选方法,鉴别了51个正调控或负调控人癌细胞生长的lncRNA。9个lncRNA中的9个均单独通过CRISPR/Cas9-介导的基因组删除和功能性回补(functionalrescue),CRISPR上调或抑制,以及基因表达图谱来验证。本发明的方法不区分lncRNA的作用机制[30],需要有详细的研究来进一步探讨被鉴别的lncRNA的功能。我们鉴别的lncRNA中有超过30%位于其它编码基因的内含子中,这些编码基因具有各种不同的生物学功能[33]。对这些lncRNA的进一步表征是具有挑战性的,因为被破坏的内含子可能对于细胞增殖具有有害的影响(例如图5b中的靶向内含子的pgRNA)。虽然在慢病毒包装和整合步骤中,由于成对gRNA的重组,CRISPRpgRNA文库可能会引起不正确的pgRNA装配,这是由于两个U6启动子和两个gRNA骨架序列之间的序列相似性,但由于重组率有限,筛选方法并没有受到影响。但是,可以通过使用不同类型的U6启动子(分别为人和鼠来源的)[34]和替代的sgRNA骨架序列来优化该方法,以进一步降低可能的慢病毒重组率。该方法还可以通过引入报告系统,从而扩展到研究除简单生长以外的感兴趣的其它表型改变。本发明的成对向导RNA筛选策略可以更广泛地应用于研究其它非编码序列,包括microRNA、顺式元件和其它未注释的元件。材料与方法细胞和试剂Huh7.5细胞来自于StanleyCohen的实验室(斯坦福大学医学院)并在加入非必需氨基酸(NEAA,Gibco)的Dulbecco’smodifiedEagle培养基(DMEM,Gibco)中培养。22RV1细胞来自于MylesBrown的实验室,并在RPMI1640培养基(Gibco)中培养。HeLa细胞来自于蒋争凡的实验室(北京大学)并保持在Dulbecco’smodifiedEagle培养基(DMEM,Gibco)中培养。所有培养基都添加了10%胎牛血清(FBS,CellMax),在5%CO2,37℃的培养箱中培养。检查所有细胞以确保它们没有支原体污染。质粒构建将人U6启动子、ccdB基因和gRNA骨架克隆到pLL3.7(Addgene,Inc.)中,替换它原有的U6启动子,构建慢病毒pgRNA-表达载体[8]。骨架-接头-U6片段被克隆到pEASY-Blunt质粒(TransGenBiotech)中。阳性和阴性对照阳性对照阳性对照的靶基因由20个基因组成,包括17个核糖体基因和3个癌症相关基因,FOXA1、HOXB13和EZH2。我们为每个阳性对照基因设计了100对gRNA,包括20对靶向启动子的(每一对中的两个gRNA之间的距离在200bp-5kb之间)gRNA,80对靶向启动子加上外显子的gRNA。阴性对照设计了三种不同类型的500个pgRNA的阴性对照。第一种类型的阴性对照(100对gRNA)由不靶向人基因组中任何位点的pgRNA组成。这些pgRNA直接由来自GeCKOv2文库[38]的现有的非靶向对照gRNA构建。第二种类型的对照(100对gRNA)由靶向AAVS1区域的pgRNA组成,AAVS1区域是基因组中的非必需区域,常常在CRISPR研究中使用,用于进行效率检验。第三种类型的阴性对照(300对gRNA)由靶向阳性对照基因的内含子的pgRNA组成。CRISPR/Cas9pgRNA文库的构建用12472对gRNA构建了一个文库,其靶向671个lncRNA。参见图3a,设计并合成含有每一对pgRNA的gRNA配对序列编码序列的137-nt的寡核苷酸(CustomArray,Inc.)。用靶向这些寡核苷酸侧翼序列的引物进行扩增,以产生与BsmBI消化的表达pgRNA的骨架同源的60bp同源序列。扩增的DNA产物通过Gibson组装方法[39]被连接到慢病毒载体中,并被转化到Trans1-T1感受态细胞(Transgen,Biotech)中以获得质粒。然后用BsmBI消化上一步获得的质粒并与BsmBI消化的骨架-Linker-U6片段相连(图4),将连接混合物转化到Trans1-T1感受态细胞(Transgen,Biotech)中,以获得最终的文库质粒(图13)。使用X-tremeGENEHPDNA转染试剂(Roche)将文库质粒与两个病毒包装质粒pVSVG和pR8.74(Addgene,Inc.)共转染到HEK293T细胞中,产生pgRNA文库的慢病毒。通过低MOI(~0.3)病毒的转导构建Huh7.5OC细胞文库,感染后72h,进行FACS筛选侵染成功的细胞。重组率计算计算转导后质粒构建体中的和细胞中染色体整合的重组率。对于质粒来说,从文库质粒扩增了整个pgRNA序列作为模板。对于细胞中的染色体整合来说,从文库基因组扩增pgRNA序列作为模板。然后将PCR产物克隆到载体中进行测序分析。分别从质粒和细胞文库随机选择80和120个克隆进行测序。CRISPR/Cas9pgRNA文库筛选将总共1.2×107个pgRNA文库细胞接种到150mm的Petri板上,设置三个重复组。收集对照组的文库细胞,并将实验组的文库细胞培养一个月,收集实验组的细胞。使用DNeasyBloodandTissuekit(Qiagen)从4×106个细胞中分离每个重复的基因组DNA。然后用靶向U6启动子和每一对的两个gRNA之间的接头的引物,通过28个循环的反应,PCR扩增(TransTaqDNAPolymeraseHighFidelity,TransGen)被整合到染色体中的gRNA-编码区域(图4和表12)。在每一管中,使用0.6μg的基因组DNA作为模板,对每一个重复进行20个PCR反应。将每一个重复的PCR产物混合在一起,并用DNAClean&Concentrator-25(ZymoResearchCorporation)纯化,然后进行深度测序分析(IlluminaHiSeq2500)。筛选的计算分析使用MAGeCK的最新版本(0.5.3),进行文库筛选后的数据分析[21]。MAGeCK算法由4步组成:标准化,pgRNA均值方差建模,pgRNA排序和lncRNA排序。通过综合分析靶向每个lncRNA的不同pgRNA的富集程度、在不同重复组的平行性、每个lncRNA有显著改变的pgRNA数量给出综合评分,得到负向筛选和正向筛选中被显著富集的基因,用于进行后续的遗传学和功能性验证。细胞增殖检测待验证的靶向阳性对照基因和lncRNA的所有pgRNA被克隆到携带有CMV启动子驱动的EGFP的慢病毒表达骨架中,并通过病毒侵染被递送到细胞中。通过FACS对EGFP+细胞的百分比进行定量。第一次定量开始于病毒感染后三天,并标记为第0天,作为标准化的对照。将给定时间点的EGFP+百分比相对于第0天对照进行标准化,来确定细胞增殖率。细胞死亡信号检测靶向负向筛选lncRNA的所有pgRNA通过慢病毒感染被递送到Huh7.5OC细胞中,被设计用于阻抑或上调lncRNA的转录水平的所有sgRNA通过瞬时转染被递送到Huh7.5细胞中。感染或转染后72小时对细胞进行FACS富集,在FACS后一天到三天进行LDH死亡信号检测。LDH染色和检测的进行如产品说明所述(CytTox96,Promega)。LDH释放数量所代表的死亡信号相对于全部裂解细胞的最大LDH活性的孔进行标准化。CRISPR抑制转录和CRISPR上调转录对于CRISPRi,将KRAB-dCas9-P2A-mCherry(Addgene#60954)质粒通过慢病毒感染递送到Huh7.5细胞中。感染3天后通过FACS富集mCherry-阳性细胞。然后将靶向负向筛选lncRNA的sgRNA通过慢病毒转染递送到稳定表达dCas9-KRAB的细胞中,然后进行细胞增殖检测和细胞致死性检测。对于CRISPRa,将三个质粒dCAS-VP64_Blast(Addgene#61425),MS2-P65-HSF1_Hygro(Addgene#61426)和携带有EGFP的针对每个正向筛选lncRNA的sgRNA通过瞬时转染递送到细胞中。转染3天后通过FACS富集EGFP阳性的细胞,然后进行细胞致死性检测。实时PCR使用RNAprepPureMicro试剂盒(TIANGEN,DP420)提取培养细胞的RNA,然后用QuantScriptRTkit(TIANGEN,KR103-03)合成cDNA。用SYBRPremixExTaqII(TaKaRa,RR820A)在LightCycler96qPCR系统上进行实时PCR。测量GAPDH转录水平作为标准化的对照。基因组DNA和文库构建的PCR扩增所使用的引物表12基因组DNA和文库构建的PCR扩增所使用的引物定量PCR的引物表13定量PCR的引物参考文献1.Barrangou,R.etal.CRISPRprovidesacquiredresistanceagainstvirusesinprokaryotes.Science315,1709-1712(2007).2.Jinek,M.etal.Aprogrammabledual-RNA-guidedDNAendonucleaseinadaptivebacterialimmunity.Science337,816-821(2012).3.Cong,L.etal.MultiplexGenomeEngineeringUsingCRISPR/CasSystems.Science339,819-823(2013).4.Mali,P.etal.RNA-guidedhumangenomeengineeringviaCas9.Science339,823-826(2013).5.Shalem,O.etal.Genome-scaleCRISPR-Cas9knockoutscreeninginhumancells.Science343,84-87(2014).6.Wang,T.etal.Identificationandcharacterizationofessentialgenesinthehumangenome.Science350,1096-1101(2015).7.Koike-Yusa,H.,Li,Y.,Tan,E.P.,Velasco-HerreraMdel,C.&Yusa,K.Genome-widerecessivegeneticscreeninginmammaliancellswithalentiviralCRISPR-guideRNAlibrary.NatBiotechnol32,267-273(2014).8.Zhou,Y.etal.High-throughputscreeningofaCRISPR/Cas9libraryforfunctionalgenomicsinhumancells.Nature509,487-491(2014).9.Canver,M.C.etal.BCL11AenhancerdissectionbyCas9-mediatedinsitusaturatingmutagenesis.Nature527,192-197(2015).10.Rajagopal,N.etal.High-throughputmappingofregulatoryDNA.NatBiotechnol34,167-174(2016).11.Korkmaz,G.etal.FunctionalgeneticscreensforenhancerelementsinthehumangenomeusingCRISPR-Cas9.NatBiotechnol34,192-198(2016).12.Shalem,O.,Sanjana,N.E.&Zhang,F.High-throughputfunctionalgenomicsusingCRISPR-Cas9.NatRevGenet(2015).13.Peng,J.,Zhou,Y.,Zhu,S.&Wei,W.High-throughputscreensinmammaliancellsusingtheCRISPR-Cas9system.FEBSJ282,2089-2096(2015).14.Han,J.etal.EfficientinvivodeletionofalargeimprintedlncRNAbyCRISPR/Cas9.RNAbiology11(2014).15.Yin,Y.etal.OpposingRolesforthelncRNAHauntandItsGenomicLocusinRegulatingHOXAGeneActivationduringEmbryonicStemCellDifferentiation.CellStemCell16,504-516(2015).16.Ren,Q.etal.ADual-ReporterSystemforReal-TimeMonitoringandHigh-throughputCRISPR/Cas9LibraryScreeningoftheHepatitisCVirus.Scientificreports5,8865(2015).17.Zheng,Q.etal.PrecisegenedeletionandreplacementusingtheCRISPR/Cas9systeminhumancells.Biotechniques57,115-124(2014).18.Du,Z.etal.IntegrativegenomicanalysesrevealclinicallyrelevantlongnoncodingRNAsinhumancancer.NatStructMolBiol20,908-913(2013).19.Hsu,P.D.etal.DNAtargetingspecificityofRNA-guidedCas9nucleases.NatBiotechnol31,827-832(2013).20.Xu,H.etal.SequencedeterminantsofimprovedCRISPRsgRNAdesign.GenomeRes25,1147-1157(2015).21.Li,W.etal.MAGeCKenablesrobustidentificationofessentialgenesfromgenome-scaleCRISPR/Cas9knockoutscreens.GenomeBiol15,554(2014).22.Subramanian,A.etal.Genesetenrichmentanalysis:aknowledge-basedapproachforinterpretinggenome-wideexpressionprofiles.ProcNatlAcadSciUSA102,15545-15550(2005).23.Cheng,A.S.etal.EZH2-mediatedconcordantrepressionofWntantagonistspromotesbeta-catenin-dependenthepatocarcinogenesis.CancerRes71,4028-4039(2011).24.Gillies,S.D.,Morrison,S.L.,Oi,V.T.&Tonegawa,S.Atissue-specifictranscriptionenhancerelementislocatedinthemajorintronofarearrangedimmunoglobulinheavychaingene.Cell33,717-728(1983).25.Xiao,X.etal.Splicesitestrength-dependentactivityandgeneticbufferingbypoly-Gruns.NatStructMolBiol16,1094-1100(2009).26.Gilbert,L.A.etal.Genome-ScaleCRISPR-MediatedControlofGeneRepressionandActivation.Cell159,647-661(2014).27.Konermann,S.etal.Genome-scaletranscriptionalactivationbyanengineeredCRISPR-Cas9complex.Nature517,583-588(2015).28.Eferl,R.&Wagner,E.F.AP-1:adouble-edgedswordintumorigenesis.Naturereviews.Cancer3,859-868(2003).29.Hatziapostolou,M.etal.AnHNF4alpha-miRNAinflammatoryfeedbackcircuitregulateshepatocellularoncogenesis.Cell147,1233-1247(2011).30.Rinn,J.L.&Chang,H.Y.GenomeregulationbylongnoncodingRNAs.AnnuRevBiochem81,145-166(2012).31.Barretina,J.etal.TheCancerCellLineEncyclopediaenablespredictivemodellingofanticancerdrugsensitivity.Nature483,603-607(2012).32.Sramkoski,R.M.etal.Anewhumanprostatecarcinomacellline,22Rv1.InVitroCellDevBiolAnim35,403-409(1999).33.Louro,R.,Smirnova,A.S.&Verjovski-Almeida,S.LongintronicnoncodingRNAtranscription:expressionnoiseorexpressionchoice?Genomics93,291-298(2009).34.Vidigal,J.A.&Ventura,A.Rapidandefficientone-stepgenerationofpairedgRNACRISPR-Cas9libraries.Naturecommunications6,8083(2015).35.Yates,A.etal.Ensembl2016.NucleicAcidsRes44,D710-716(2016).36.Smyth,G.K.Linearmodelsandempiricalbayesmethodsforassessingdifferentialexpressioninmicroarrayexperiments.StatApplGenetMolBiol3,Article3(2004).37.Wang,T.,Wei,J.J.,Sabatini,D.M.&Lander,E.S.GeneticscreensinhumancellsusingtheCRISPR-Cas9system.Science343,80-84(2014).38.Sanjana,N.E.,Shalem,O.&Zhang,F.Improvedvectorsandgenome-widelibrariesforCRISPRscreening.NatMethods11,783-784(2014).39.Gibson,D.G.EnzymaticassemblyofoverlappingDNAfragments.MethodsEnzymol498,349-361(2011).40.Anders,S.&Huber,W.Differentialexpressionanalysisforsequencecountdata.GenomeBiol11,R106(2010).41.Kim,D.etal.TopHat2:accuratealignmentoftranscriptomesinthepresenceofinsertions,deletionsandgenefusions.GenomeBiol14,R36(2013).42.Anders,S.,Pyl,P.T.&Huber,W.HTSeq--aPythonframeworktoworkwithhigh-throughputsequencingdata.Bioinformatics31,166-169(2015).43.Love,M.I.,Huber,W.&Anders,S.ModeratedestimationoffoldchangeanddispersionforRNA-seqdatawithDESeq2.GenomeBiol15,550(2014).44.Taylor,B.S.etal.Integrativegenomicprofilingofhumanprostatecancer.Cancercell18,11-22(2010).45.CancerGenomeAtlasResearch,N.Comprehensivegenomiccharacterizationdefineshumanglioblastomagenesandcorepathways.Nature455,1061-1068(2008).46.Partensky,F.&Garczarek,L.Microbiology:Armsraceinadropofseawater.Nature474,582-583(2011).47.CancerGenomeAtlasResearch,N.Comprehensivegenomiccharacterizationofsquamouscelllungcancers.Nature489,519-525(2012).48.Li,J.etal.TANRIC:AnInteractiveOpenPlatformtoExploretheFunctionoflncRNAsinCancer.CancerRes75,3728-3737(2015).49.Wilks,C.etal.TheCancerGenomicsHub(CGHub):overcomingcancerthroughthepoweroftorrentialdata.Database(Oxford)2014(2014).50.Trapnell,C.etal.DifferentialgeneandtranscriptexpressionanalysisofRNA-seqexperimentswithTopHatandCufflinks.NatProtoc7,562-578(2012).51.Alexa,A.,Rahnenfuhrer,J.&Lengauer,T.ImprovedscoringoffunctionalgroupsfromgeneexpressiondatabydecorrelatingGOgraphstructure.Bioinformatics22,1600-1607(2006).当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1