苯并菲炔联苯炔苯桥连苝酰亚胺二酯二元化合物的合成方法与流程
本发明涉及一种苯并菲炔联苯炔苯桥连苝酰亚胺二酯二元化合物的合成方法。
背景技术:
柱状相盘状液晶材料处于液晶相时,由于盘状核之间较强的π-π电子相互作用,易自组装成有序的分子柱。从而使得沿分子柱的轴向具有较高的载流子迁移率,可以作为光电子器件的活性层和修饰层使用。
苯并菲类液晶材料制备方法简单、产率较高、易于提纯,紫外光区有较强吸收,根据报道其可以作为有机发光器件的空穴传输层、发光中心和空穴传输层等,是目前研究地最为广泛的盘状液晶材料。尤其是在光学,光电学和电子学具有广泛应用前景。而苝酰亚胺类盘状液晶材料是一类具有特殊稠环结构的化合物,具有良好的光、热稳定性和抗氧化性,同时拥有较宽的光谱吸收范围,以及高荧光量子产率,低Stokes位移等优良的光学性质,且由于苝核呈缺电子体系性质,具有较高的电子亲和能,这使得苝酰亚胺类衍生物成为一种典型的n型有机半导体材料,因此在有机光伏领域得到了广泛研究。
技术实现要素:
本发明的目的是提供一种苯并菲炔联苯炔苯桥连苝酰亚胺二酯二元化合物的合成。
具体步骤为:
(1)将邻二己氧基苯和2-己氧基苯酚在三氯化铁的氧化作用下发生偶联反应生成单羟基五己氧基苯并菲;然后,以单羟基五己氧基苯并菲为原料合成含有炔联苯炔基五烷氧基苯并菲衍生物(化合物8)。
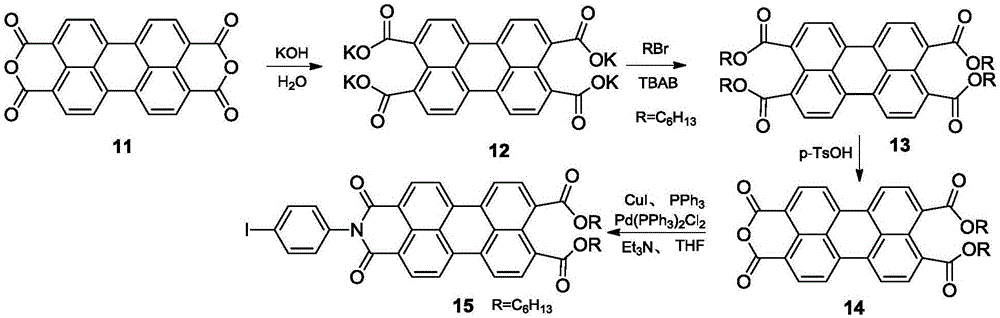
(2)以苝二酸酐为原料合成苝酸酐二酯;然后苝酸酐二酯与4-碘苯胺进行酰胺化得到苝酰亚胺二酯(化合物15)。
(3)将含有炔联苯炔基五烷氧基苯并菲衍生物与苝酰亚胺二酯中间体偶联反应得到苯并菲炔联苯炔苯桥连苝酰亚胺二酯二元化合物(化合物16)。
本发明设计并合成了以烷氧基苯并菲单元为电子给体(Donor),苝酰亚胺单元为电子受体(Acceptor)的有D-B-A型结构的二元化合物,这类化合物既具有分子内光诱导电子转移的性质,又可能具有柱状相盘状液晶的性质。预期得到具有使电子和空穴分别沿盘状液晶受体单元的分子柱和给体单元的分子柱定向迁移的性质,从而减少载流子的损失,极有希望提高有机光伏器件的性能。
附图说明
图1为本发明含有炔苯炔基五烷氧基苯并菲衍生物(化合物8)的合成路线图。
图中标记为:1-化合物1,2-化合物2,3-化合物3,4-化合物4,5-化合物5,6-化合物6,7-化合物7,8-化合物8。
图2为本发明苝酰亚胺二酯(化合物15)的合成路线图。
图中标记为:11-化合物11,12-化合物12,13-化合物13,14-化合物14,15-化合物15。
图3为本发明苯并菲炔联苯炔苯桥连苝酰亚胺二酯二元化合物(化合物16)的合成路线图。
图中标记为:8-化合物8,15-化合物15,16-化合物16。
具体实施方式
实施例:
实施例中所使用的化学试剂和溶剂均为分析纯。
(1)邻己氧基苯酚(化合物1)的合成
将邻苯二酚(20g,0.1816mol)、1-溴代正己烷(89.9g,0.5448mol)、无水碳酸钾(73.5g,0.5448mol)、碘化钾(6.0g)和无水乙醇(250mL)混合,将反应液温度升至85℃回流60小时后,将反应的混合物冷却到室温抽滤,用乙醇(150mL)洗涤滤饼,抽滤,重复两次。滤液旋蒸后,再减压蒸馏收集142-155℃(0.5mmHg)的馏分,得到化合物1为无色油状液体(48g),产率96%;IR(KBr)νmax(cm-1):3540,3050,2930,2850,1800,1600,1500,1360,1260.1H NMR(300MHz,CDCl3)δ:6.89(s,4H),3.99(t,J=6.9Hz,4H),1.83-1.76(m,J=6.9Hz,4H),1.49-1.31(m,12H),0.9(t,J=6.9Hz,6H)。
(2)1,2-二己氧基苯(化合物2)的合成
将邻苯二酚(50g,0.45mol)、1-溴代正己烷(75g,0.45mol)、无水碳酸钾(100g,0.725mol)、碘化钾(3.8g)和无水乙醇(500mL)混合,将混合液温度升至85℃回流12小时后,将反应的混合物冷却到室温后抽滤,用乙醇(150mL)洗涤滤饼,抽滤,重复两次。滤液旋蒸后,在减压蒸馏收集140-143℃(0.5mmHg)的馏分,得到化合物2为无色油状液体(25.5g),产率29%;IR(KBr)νmax(cm-1):3060,2930,2860,1590,1470,1500,1390,1250.1H NMR(300MHz,CDCl3)δ:6.95-6.82(m,4H),5.68(s,1H),4.02(t,J=6.9Hz,2H),1.83-1.76(m,2H),1.48-1.31(m,6H),0.91(t,J=6.9Hz,3H)。
(3)2-羟基-3,6,7,10,11-五己氧基苯并菲(化合物3)的合成
将无水三氯化铁(12.96g,79.2mol)溶于二氯甲烷(80mL)和硝基甲烷(8mL)中。化合物1(4.44g,16mmol)和化合物2(1.56g,8mmol)溶于二氯甲烷(60mL),在0℃时,将化合物1和化合物2的混合溶液滴加至上述无水三氯化铁的二氯甲烷和硝基甲烷溶液中,30分钟滴加完毕。将反应的混合物保持在0℃-3℃范围内继续反应3小时后,加入50mL甲醇和25mL水终止反应。反应的混合物用15mL浓度为2mol/L的稀盐酸洗涤两次,用15mL饱和食盐水洗涤一次。分出有机层,加入无水硫酸钠干燥除水后,减压旋蒸出溶剂,得到的粗产物用硅胶色谱柱提纯(淋洗液为:乙酸乙酯/石油醚体积比为1:30-1:15),得到2.14g白色固体即化合物3,产率为36%。47.0-50.0℃;IR(KBr)νmax(cm-1):3734,3446,2957,2859,2360,2340,1618,1518,1469,1265,1171,1105,860,727,650,516;1H NMR(400MHz,CDCl3)δ:7.96(s,1H),7.83(s,3H),7.82(s,1H),7.77(s,1H),5.91(s,1H),4.31-4.19(m,10H),1.98-1.90(m,10H),1.59-1.40(m,30H),0.96-0.92(m,15H)。
(4)2-三氟甲磺酸酯-3,6,7,10,11-五己氧基苯并菲(化合物4)的合成
在氮气保护下,取化合物3(0.65g,0.87mmol)置于干燥三口瓶,并加入6mL吡啶溶解,冰水浴下搅拌15分钟。在氮气保护下三氟甲磺酸酐(0.30g,1.05mmol)逐滴加入三口瓶,滴加完成后15分钟撤去冰浴。氮气保护,室温下反应5小时。反应液用10mL质量百分比浓度5%的稀盐酸和20mL二氯甲烷萃取,收集有机层,减压蒸馏得到粗产物。粗产物用体积比为1:4的二氯甲烷-甲醇重结晶,干燥后得0.67g浅褐色固体即化合物4,产率87.56%;Mp:166.5-167℃;IR(KBr)νmax(cm-1):3734,3446,2957,2859,2360,2340,1618,1518,1469,1265,1171,1105,860,727,650,516;1H NMR(500MHz,CDCl3)δ8.19(s,1H),7.87(s,1H),7.82(s,1H),7.80(s,2H),7.71(s,1H),4.29-4.21(m,10H),1.99-1.91(m,10H),1.63-1.55(m,10H),1.41(m,20H),0.96-0.92(m,15H)。
(5)2-(3-甲基-3-羟基-1-丁炔基)-3,6,7,10,11-五己氧基苯并菲(化合物5)的合成
氮气保护下,取0.67g化合物4(0.76mmol)、三苯基膦(0.0203g,0.076mmol)0.0148g碘化亚铜(0.076mmol)和二氯化二(三苯基磷)合钯(II)(0.0540g,0.076mmol)加入三口瓶,然后加入15mL四氢呋喃、5mL三乙胺搅拌溶解,然后加入2-甲基-3-丁炔-2-醇(0.098g,1.15mmol)氮气保护,80℃搅拌回流过夜。反应液用80mL质量百分比浓度为5%氯化铵溶液/20mL二氯甲烷萃取,收集有机层,减压蒸馏得粗产物。粗产物用硅胶色谱柱进行提纯,洗脱液比例乙酸乙酯:石油醚体积比为10:1,得到0.43g化合物5,产率87.56%;Mp:131-132℃;IR(KBr)νmax(cm-1):3735,3448,2928,2360,1615,1517,1434,1264,1175,959,837,750,616;1HNMR(400MHz,CDCl3)δ8.46(s,1H),7.86(d,J=7.2Hz,2H),7.81(s,2H),7.74(s,1H),4.23(m,10H),2.17(s,1H),1.94(t,J=5.2Hz,10H),1.72(s,6H),1.63-1.55(m,10H),1.41(m,20H),0.94(m,15H)。
(6)2-炔基-3,6,7,10,11-五己氧基苯并菲(化合物6)的合成
氮气保护下,将化合物5(0.43g,0.53mmol)、氢氧化钾(0.27g,4.77mmol)和25ml甲苯加入烧瓶,110℃搅拌回流2小时。反应液用水和二氯甲烷萃取,收集有机层,减压蒸馏得粗产物。粗产物用硅胶色谱柱进行提纯,洗脱液比例乙酸乙酯:石油醚体积比为1:30,得0.25g浅黄色固体即化合物6,产率61.4%;Mp:124-126℃;IR(KBr)νmax(cm-1):3445,3315,2956,2858,2360,2101,1614,1516,1434,1263,1171,1041,929,834,727,636;1HNMR(400MHz,CDCl3)δ8.46(s,1H),7.86(d,J=7.2Hz,2H),7.81(s,2H),7.74(s,1H),4.23(m,10H),4.0(s,1H),1.94(t,J=5.2Hz,10H),1.63-1.55(m,10H),1.41(m,20H),0.94(m,15H)。
(7)2-(3-甲基-3-羟基-1-丁炔基-4-联苯)乙炔基-3,6,7,10,11-五己氧基苯并菲(化合物7)的合成
氮气保护下,将事先干燥处理过4-(4-碘联苯)-2-甲基-3-丁炔-2-醇(0.32g,0.88mmol)、化合物6(0.66g,0.88mmol)、三苯基膦(23.0mg,0.09mmol),碘化亚铜(8.4mg,0.04mmol)和双(三苯基膦)二氯化钯(Ⅱ)(61.5mg,0.09mmol)加入干燥的三口瓶中,抽真空,充氮气,重复三次。然后在正压保护下,分别加入四氢呋喃(18mL)和三乙胺(6mL),升温80℃,搅拌回流,反应过夜。冷却至室温,反应液用80mL质量百分比浓度为5%氯化铵溶液/20mL二氯乙烷萃取三遍,收集有机层,饱和食盐水洗涤,无水硫酸钠干燥,旋蒸除去溶剂。粗产物用硅胶色谱柱提纯,得到白色固体即化合物7(0.70g,产率81%),Mp:67-68℃;1H NMR(400MHz,CDCl3)δ8.58(s,1H),7.92(s,1H),7.87(s,1H),7.80(d,J=6.8Hz,2H),7.70(d,J=8.0Hz,2H),7.60(dd,J=12.8,8.8Hz,4H),7.52(d,J=8.0Hz,2H),4.34-4.20(m,6H),2.10-2.06(m,3H),2.05-1.90(m,10H),1.65(m,5H),1.49-1.34(m,7H),1.25(s,1H),0.95(m,5H)。
(8)2-(4-乙炔基-4-联苯)乙炔基-3,6,7,10,11-五己氧基苯并菲(化合物8)的合成
氮气保护下将干燥处理过的化合物7(0.70g,0.71mmol)、氢氧化钾(0.15g,2.27mmol)加入干燥的烧瓶,正压保护下,加入甲苯(50mL)。升温,搅拌回流2小时。冷却至室温,20mL浓度为2mol/L的盐酸/20mL二氯乙烷萃取三遍,收集有机层,饱和食盐水洗涤,无水硫酸钠干燥,旋蒸除去溶剂。粗产物用硅胶色谱柱提纯,得到白色固体即化合物8(0.52g,产率79%),Mp:181-182℃;IR(KBr)νmax(cm-1):3466,2982,2360,1651,1579,1481,1370,1272,1157,1097,962,804,749,684,572;1H NMR(400MHz,CDCl3):δ8.56(s,1H),7.88(d,J=12.8Hz,2H),7.79(d,J=8.8Hz,3H),7.56(d,J=8.0Hz,4H),7.43(d,J=8.0Hz,4H),4.32-4.20(m,10H),2.05-1.91(m,11H),1.73-1.52(m,16H),1.41(ms,20H),0.95(m,15H)。
(9)苝-3,4,9,10-四甲酸四己酯(化合物13)的合成
将苝-3,4,9,10-四甲酸二酐(化合物11)(3.5g,8.92mmol)加到氢氧化钾水溶液(2.5%,400mL)中,在70℃反应1.5小时。冷却至室温,抽滤,调节滤液pH=7,得到苝四甲酸钾(化合物12)的水溶液。将1-溴己烷(14.72g,89.2mmol)、四丁基溴化铵(1.45g,4.5mmol)加到滤液中,在100℃反应6小时。冷却至室温,用体积比为1:3的水/二氯甲烷萃取,收集有机层,饱和食盐水洗涤,无水硫酸钠干燥,减压蒸馏除去溶剂。粗产物用体积比为1:1的二氯甲烷/乙醇重结晶抽滤,减压过滤得到黄色粉末即化合物13(6.51g,产率95%);Mp>300℃;IR(KBr)νmax(cm-1):3430,2930(C-H),2850(C-H),1730(C=O),1630,1460,1400,1270(C-O),1170,1000,747;1HNMR(500MHz,CDCl3)δ:8.15(d,J=8.0Hz,2H),8.09(d,J=8.0Hz,2H),7.98-7.93(m,4H),4.35(t,J=7.0Hz,8H),1.86-1.80(m,8H),1.50-1.37(m,24H),0.93(t,J=7.0Hz,12H)。
(10)苝-3,4-二甲酸酐-9,10-二甲酸二己酯(化合物14)的合成
将化合物13(6.51g,8.51mmol)完全溶于甲苯(3.6ml)和正庚烷(18.0ml)中后,加入对甲苯磺酸(1.54g,8.95mmol),95℃反应5小时,冰水冷却后抽滤,滤饼用体积比为1:4的二氯甲烷/甲醇重结晶,冰水冷却后抽滤,重复两次,得到红色固体即化合物去14(3.88g,产率79%);Mp>300℃;IR(KBr)νmax(cm-1):3430,2920(C-H),2850(C-H),1730(C=O),1630,1290(C-O),1150,1010,857,805,737;1HNMR(500MHz,CDCl3)δ:8.15(d,J=8.0Hz,2H),8.09(d,J=8.0Hz,2H),7.98-7.93(m,4H),4.35(t,J=7.0Hz,4H),1.86-1.80(m,4H),1.50-1.37(m,12H),0.93(t,J=7.0Hz,6H)。
(11)苝-N-对碘苯炔-3,4-二甲酰亚胺-9,10-二甲酸二己酯(化合物15)的合成
氮气保护下,向烧瓶中加入干燥过的化合物14(2.00g,3.46mmol)、4-碘苯胺(3.03g,13.83mmol)、无水醋酸锌(0.32g,1.73mmol)和咪唑(20.00g),抽真空充氮气,重复三次。搅拌升温150℃,反应过夜,冷却至室温。反应物用体积比为1:3的水/二氯甲烷萃取,收集有机层,饱和食盐水洗涤,无水硫酸钠干燥,旋蒸除去溶剂。用体积比为1:1的二氯甲烷/乙醇重结晶,得到橙红色固体即化合物15(2.65g,产率98%)。Mp:254-255℃;IR(KBr)νmax(cm-1):3450,2956,2856,1709,1667,1596,1511,1469,1366,1274,1177,1067,1012,961,809,778;1H NMR(400MHz,CDCl3)δ8.57(d,J=8.0Hz,2H),8.35(dd,J=8.2,4.5Hz,4H),8.07(d,J=8.0Hz,2H),7.90(d,J=8.4Hz,2H),7.14(d,J=8.4Hz,2H),4.35(t,J=6.8Hz,4H),1.83(q,J=7.2Hz,4H),1.46(m,4H),1.36(m,8H),0.97-0.88(m,6H)。
(12)苯并菲炔联苯炔苯桥连苝二元化合物(化合物16)的合成
氮气保护下,将事先干燥处理过的化合物8(0.45g,0.60mmol)、化合物15(0.49g,0.63mmol)、三苯基膦(15.7mg,0.06mmol)、碘化亚铜(11.4mg,0.06mmol)和双(三苯基膦)二氯化钯(Ⅱ)(41.9mg,0.06mmol)加入干燥的三口瓶中,架好装置抽真空,充氮气,重复三次。然后在正压保护下,分别加入四氢呋喃(14mL)和三乙胺(3.5mL)。升温80℃,避光搅拌回流,反应过夜。冷却至室温,反应液用80mL质量百分比浓度为5%氯化铵溶液/20mL三氯甲烷萃取三遍,收集有机层,饱和食盐水洗涤,无水硫酸钠干燥,旋蒸除去溶剂。粗产物用硅胶色谱柱提纯,得到白色固体即化合物16(0.69g,产率82%),Mp:241.8-243.6℃;IR(KBr)νmax(cm-1):3446,2953,2928,2857,2360,2340,1705,1668,1593,1511,1468,1432,1360,1296,1263,1171,1064,1036,879,839,806,748,726,669,599.1H NMR(500MHz,CDCl3)δ8.61(d,J=8.0Hz,2H),8.45(s,1H),8.37(d,J=8.0Hz,2H),8.31(d,J=8.0Hz,2H),8.06(d,J=8.0Hz,2H),7.82-7.77(m,3H),7.76(s,1H),7.65(s,2H),7.62(s,1H),7.47-7.43(m,2H),4.35(t,J=6.8Hz,4H),4.30-4.16(m,10H),2.07-1.89(m,10H),1.87-1.78(m,4H),1.69(m,2H),1.52-1.33(m,30H),1.00-0.90(m,21H)。
- 还没有人留言评论。精彩留言会获得点赞!