基于高通量测序技术的小麦病原微生物检测方法及其应用与流程
本申请涉及小麦病原微生物检测领域,特别是涉及一种基于高通量测序技术的小麦病原微生物检测方法及其应用。
背景技术:
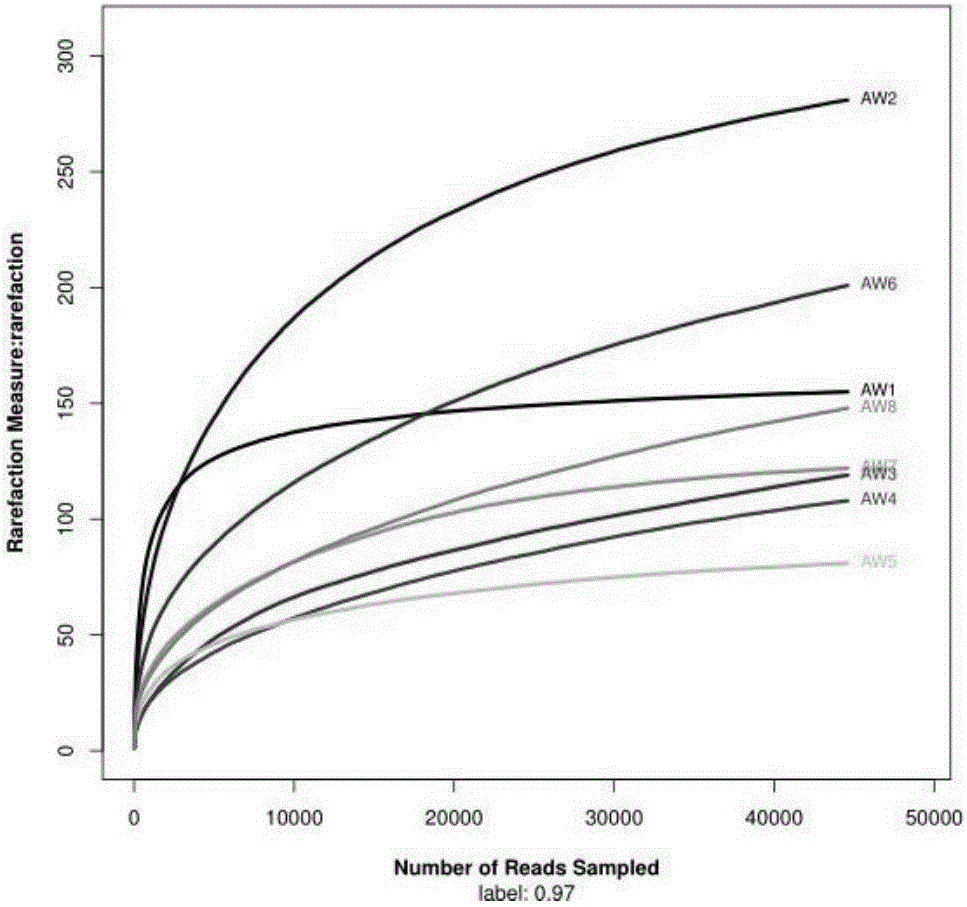
:中国是最大的小麦进口国,小麦进口量呈逐年增长态势。小麦上携带的病原真菌通过贸易方式传入、定殖会对我国的农业生产安全带来很大的风险。目前,全世界小麦上病害有200多种,其中可以通过输入小麦传播的有25种病原物,4种检疫性病原物,此外还有一些潜在风险的病原物,这些种传病害经由贸易途径传带,是植物检疫和农业主管部门重点关注的对象。目前,对小麦病原微生物的检测主要有形态学、生化学、血清学和分子生物学方法。这些检测手段大多都是基于对病原菌分离培养后的分析。以世界范围内广泛关注的检疫性病原真菌—小麦印度腥黑穗病菌检疫鉴定的国际标准(ISPM27.2006Annex4:TilletiaindicaMitra(2004-014))和国家标准(GB/T28080-2011)为例,样品前处理需先将冬孢子洗涤下来,再进行形态学和分子生物学的鉴定。正如已被分离培养的微生物只是微乎其微的极少部分,被人们分离培养并认知的病原微生物同样极少。专性寄生、培养条件苛刻、生长速度慢、在群体中丰度低、特殊生态要求等影响了病原微生物的分离培养,而这些限制就对研究病原菌的分类、多样性、致病机理、检测方法、风险分析及防控措施提出了挑战。在植物检疫领域,检疫性物种及其近似种的区分、重要有害生物的漏检、检疫处理措施的制定等都受到微生物不能分离培养的制约,这一限制性因素对检测方法的制定和结果的判定又有着更特殊、更重要的意义。因此,亟需对小麦上这些因不能分离培养的病原微生物进行认识和分析,评估其有害性和风险性,在传统风险分析的基础上,补充提出重点关注的有害生物名单,应对国际植物及植物产品贸易中的技术壁垒。高通量测序技术具有高效、快速、廉价并全面分析复杂的核苷酸的功能。目前,高通量测试技术已经应用于临床医学,用以对疾病或用药相关基因的突变进行全面检测,进而指导用药和预后。但是,在小麦病原微生物检测方面,尚没有利用高通量测序技术的高通量优势进行病原微生物检测的研究和报道。技术实现要素:本申请的目的是提供一种基于高通量测序技术的小麦病原微生物检测方法及其应用。为了实现上述目的,本申请采用了以下技术方案:本申请一方面公开了基于高通量测序技术的小麦病原微生物检测方法,包括以下步骤,1)提取待检小麦样品或小麦筛下物样品的基因组DNA;2)采用真菌ITS区通用引物对步骤1)的基因组DNA进行PCR扩增建库;3)对步骤2)的PCR扩增产物进行高通量测序;4)将高通量测序结果根据97%以上相似性进行聚类分析,根据获得的分类单元分析取待检小麦样品或小麦筛下物样品的病原微生物群落。需要说明的是,本申请的检测方法,创造性的将高通量测序技术引入小麦病原微生物的检测,通过对基因组DNA进行ITS区测序和聚类分析,可以全面掌握小麦上所携带的几乎所有病原微生物种类。本申请的检测方法为全面的认识和分析小麦病原微生物奠定了基础,进而为评估其有害性和风险性,在传统风险分析的基础上,补充提出重点关注的有害生物名单,应对国际植物及植物产品贸易中的技术壁垒提供了有力的科学依据。优选的,本申请的小麦病原微生物检测方法,还包括将步骤4)分析获得的病原微生物群落在目或属的分类水平上,将其分为优势类群、常见类群、次常见类群和稀有类群四大类;优势类群为比例大于或等于总病原微生物群落10%的真菌群落;常见类群为比例大于或等于总病原微生物群落1%,并小于10%的真菌群落;次常见类群为比例大于或等于总病原微生物群落0.5%,并小于1%的真菌群落;稀有类群为比例小于总病原微生物群落0.5%的真菌群落。需要说明的是,在本申请的一种实现方式中,对进境小麦分析获得了622个分类单元(缩写OTU),这622个OTU分属于5个门64个目201个属的真菌;为了便于分析和认识,本申请创造性的提出将分析获得的病原微生物群落分别在不同分类水平上将其分为优势类群、常见类群、次常见类群和稀有类群四大类,以便于深入研究。本申请的另一面公开了本申请的小麦病原微生物检测方法在进境小麦样品检测中的应用。本申请的另一面公开了高通量测序技术在进境澳大利亚小麦病原微生物检测中的应用,包括采用本申请的小麦病原微生物检测方法对进境澳大利亚小麦或其筛下物中所携带的病原微生物群落进行全面分析和检测。需要说明的是,本申请的一种实现方式中,对进境澳大利亚小麦进行了病原微生物检测,分析获得了分属于5个门64个目201个属的真菌,为全面了解进境澳大利亚小麦上所携带的病原微生物打下了坚实的基础。由于采用以上技术方案,本申请的有益效果在于:本申请的基于高通量测序技术的小麦病原微生物检测方法,利用高通量测序技术,对小麦或小麦筛下物的基因组DNA进行测序,并通过生物信息学分析得到OTU,进而全面的分析出小麦所携带的病原真菌情况,进而为评估其有害性和风险性奠定了基础。本申请的检测方法特别适用于进出境检验检疫等部门,以便于全面的了解小麦病菌携带情况,进而突破或制定出相应的技术壁垒,保障小麦生产和贸易的安全。附图说明图1是本申请实施例中8个样本的真菌稀释曲线;图2是本申请实施例中8个样本的Shannon-Wiener曲线。具体实施方式本申请的关键在于将高通量测序技术应用于小麦病原微生物的检测,基于高通量测序技术,通过对测序结果进行生物信息学分析,可以全面的了解小麦所携带的病原微生物情况,为评估其有害性和风险性,补充提出重点关注的有害生物名单,应对国际技术壁垒提供了有力的科学依据。需要说明的是,过去我们对病原微生物的学习和认识多从分类的角度入手,研究某一种病原物的分类、形态特征、寄主范围、危害性状等。而在植物病理学家、植物保护和植物检疫工作人员鉴定病原种类时,通常以某一特定感病植株的分析入手。前者侧重分类,是由点及面,并且主要是方法学层面;后者侧重性状,是由表及里,主要是系统论层面。本申请借鉴环境群体微生物生态学的思路,将一植物上的所有病原微生物视为一个群体,对其多样性进行研究可以知晓其致病微生物的种类和数量,分析不同病原菌之间的生态关系是否对侵染、定殖、致病产生影响。因此,本申请的小麦病原微生物检测方法,对掌握进口小麦或小麦产品上病原物的多样性,对其进行风险评估,进而提出和完善重点关注有害生物名单,甚至突破技术壁垒,制订新的技术壁垒等,都具有重大意义。还需要说明的是,在研究植物病原微生物多样性时会遇到两个问题:一是微生物多样性的特殊性,不同产地、不同品种的同一种植物上的病原种类和数量都不同。某一特定的寄主上的病原物多样性受内、外多种因素影响,内在因素有种子健康情况、植物生长发育阶段、生长健康程度等,外界因素有自然条件,如水分、空气、温度等环境因子,某一病原物的近似寄主,田间管理措施,如杀菌剂的使用、病原物其他寄主的清除、灌溉情况等。第二个问题是,正如已被分离培养的微生物只是微乎其微的极少部分,目前,已知细菌4760种,估计总种数40000种,被分离培养的只有约12%;已知真菌72000,估计总种数1.5×106种,被分离培养的只有约5%,被人们分离培养并认知的病原微生物同样极少。专性寄生、培养条件苛刻、生长速度慢、在群体中的丰度低、特殊生态关系等影响了病原微生物分离培养,而这些限制就对研究病原菌多样性的认识提出了挑战。在植物检疫领域,检疫性物种及其近似种的区分、重要有害生物的漏检、检疫处理措施的制定等都受到微生物不能分离培养的制约,这一限制性因素对检测方法的制定和结果的判定又有着更特殊、更重要的意义。而本申请的小麦病原微生物检测方法,完全不受分离培养的制约,并且能够更为全面的分析获得小麦所携带的几乎所有病原微生物的信息。这对进出境小麦的检验检疫具有重大意义。下面通过具体实施例和附图对本申请作进一步详细说明。以下实施例和附图仅对本申请进行进一步说明,不应理解为对本申请的限制。实施例本例以进境澳大利亚小麦为研究对象,样品来源为,2015年5-7月从澳大利亚进境的小麦种子及其筛下物,分别采集不同船舱的混合样品500g,具体的,编号AW1为2015年5月澳大利亚进境小麦,编号AW2为2015年5月澳大利亚进境小麦筛下物,编号AW3为2015年7月澳大利亚进境小麦,编号AW4为2015年7月澳大利亚进境小麦筛下物,编号AW5为2015年10月澳大利亚进境小麦,编号AW6为2015年10月澳大利亚进境小麦筛下物,编号AW7为2015年11月澳大利亚进境小麦,编号AW8为2015年11月澳大利亚进境小麦筛下物。一、小麦病原微生物检测方法本例的小麦病原微生物检测方法具体如下:(1)澳大利亚进境的小麦种子及其筛下物基因组DNA提取本例采用SoilDNAKit(D5625-01)对8份样品进行基因组DNA提取。具体的对500g小麦种子进行常规检疫的过筛,称取过筛后的小麦种子及其对应的筛下物各1g进行基因组DNA提取。基因组DNA提取的具体步骤详见说明书。获得基因组DNA后采用NanoDrop2000对基因组DNA进行浓度检测。(2)PCR扩增和高通量测序根据NanoDropND-2000荧光分光光度计测得的DNA浓度计算,取适量体积、总量约2μg的基因组DNA样品,寄送美吉生物技术有限公司,由生物技术公司完成PCR扩增建库和高通量测序。其中,PCR扩增采用真菌ITS区通用引物。测序分析过程在IlluminaMiseq第二代高通量测序平台上进行。测序结果已上传至NCBI的SRA数据库,编号SRP077547。(3)测序结果生物信息学分析Miseq测序得到的PEreads首先根据overlap关系进行拼接,同时对序列质量进行质控和过滤,区分样本后进行OTU聚类分析和物种分类学分析。其中,聚类分析使用upars方法在97%的相似水平下进行OTU聚类。采用RDPclassifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析。并分别在各个分类水平:门、目、属,统计各样本的群落组成。此外,本例进一步在属的水平上对小麦种子与筛下物的结果进行比较;并在属的水平上将AW1与AW2数据整合、AW3与AW4数据整合、AW5与AW6数据整合、AW7与AW8数据整合,以验证群落组成分析结果。(4)数据的合理性论证采用对测序序列进行随机抽样的方法,以抽到的序列数与它们所代表OTU的数构建曲线,即稀释性曲线(RarefactionCurve)。稀释曲线是利用克隆文库中已知的各种系型的相对比例来计算抽取n条克隆时出现种系型数量的期望值,根据一组n值与其相对应的种系型数量的期望值做出曲线,当曲线趋向平坦时,说明测序数据量合理。本例中,各种系型即OUT单元,n小于库容;本例在97%的相似水平下绘制稀释曲线。本例的软件,使用97%相似度的OTU,利用mothur做rarefaction分析,利用R语言工具制作曲线图。(5)Shannon-Wiener曲线Shannon-Wiener曲线是反映样本中微生物多样性的指数,本例利用各样本的测序量在不同测序深度时的微生物多样性指数构建曲线,以此反映各样本在不同测序数量时的微生物多样性。二、结果及分析(1)基因组DNA浓度采用NanoDrop2000对提取的8份基因组DNA浓度检测结果显示,AW1、AW2、AW3、AW4、AW5、AW、AW7、AW8的浓度依序为210ng/μL、163ng/μL、228ng/μL、180ng/μL、209ng/μL、175ng/μL、220ng/μL、178ng/μL。8份基因组DNA各取约15μL用于高通量测序检测。(2)数据的合理性论证在97%的相似水平下绘制的稀释曲线如图1所示,图中,曲线末端由上至下依序为样品AW2、AW6、AW1、AW8、AW7、AW3、AW4、AW5的曲线。结果显示,当AW1-AW8每个样品随机抽取的条带数达到4500条时,8份样品的OUT单元和多样性指数稀释曲线均已趋于平缓,说明本例的取样基本合理,能真实地反映小麦样本的真菌群落组成及多样性变化。(3)Shannon-Wiener曲线本例的Shannon-Wiener曲线如图2所示,图中,曲线末端由上至下依序为样品AW1、AW2、AW6、AW8、AW7、AW5、AW4、AW3的曲线。结果显示,AW1-AW8样品曲线趋向平坦,可见,其测序数据量足够大,可以反映样本中绝大多数的微生物信息。(4)真菌群落结构分析本例对8个基因组DNA进行了高通量测序,每个样品测序获得44604条序列。本例将8个基因组DNA的测序结果和OTU单元进行整合,共获得622个OTU,分属于5个门、64个目、201个属。不同样品间OTU种类数目各不相同:样品AW1的OTU数为155,样品AW2的OTU数为281,样品AW3的OTU数为119,样品AW4的OTU数为108,样品AW5的OTU数为81,样品AW6的OTU数为201,样品AW7的OTU数为122,AW8的OTU数为148。本例进一步的,本例分别在门、目和属三个水平对真菌菌落进行分类。结果如下:在门的分类水平上将真菌群落分为优势类群和稀有类群两大类。优势类群为比例超过99%的真菌群落,包括子囊菌和担子菌;稀有类群指那些比例小于1%的菌落,包括壶菌门Chytridiomycota、接合菌门Zygomycota。结果如表1所示。表1门的水平上澳大利亚小麦8个样本的OUT数目门AW1AW2AW3AW4AW5AW6AW7AW8Ascomycota2861634915359617233216616496321524388Basidiomycota15507957685274385312183280911171220210Chytridiomycota00110000Fungi_unclassified429112972125516296692Zygomycota5211660184(5)各分类水平的群落组成分析在目的分类水平上将真菌群落分为优势类群、常见类群、次常见类群和稀有类群四大类。优势类群为比例超过10%的真菌群落,包括Tremellales(银耳目)占32.0574%和Pleosporales(假球壳目)占34.9728%;常见类群指那些比例小于10%,大于或等于1%,但在大多数样品中均出现的真菌群落,包括Capnodiales占7.2942%、Cystofilobasidiales占3.6160%、Fungi_unclassified占8.5757%、Hypocreales占3.4162%、Tremellomycetes占比4.6574%、Trichosphaeriales占1.3457%。次常见类群指那些比例大于或等于0.5%,小于1%的真菌群落,包括Dothideales(座囊菌目)占比0.7202%、Eurotiales(散囊菌目)占比0.7483%、Sporidiobolales(锁掷酵母目)占比0.9654%。稀有类群指比例小于0.5%的真菌群落,包括Archaeorhizomycetales、Agaricales、Agaricomycetes、Agaricostilbales、Atheliales、Auriculariales、Boletales、Cantharellales、Cercozoa、Chaetothyriales、Corticiales、Cystobasidiales、Dothideomycetes、Dothideomycetes、Erythrobasidiales、Eurotiomycetes、Eurotiomycetes、Exobasidiomycetes、Exobasidiomycetes、Filobasidiales、Geminibasidiales、Glomerellales、Incertae、Leotiomycetes、Leucosporidiales、Malasseziales、Microbotryomycetes、Mortierellales、Olpidiales、Orbiliales、Xylariales、Ustilaginales、Thelephorales、Thelebolales、Sordariomycetes、Sordariales、Saccharomycetales、Rhytismatales、Polyporales、Pezizales等40种真菌类群,以及Ascomycota(子囊菌属)占比0.2396%、Basidiomycota占比0.1317%、Erysiphales(白粉菌目)占比0.2186%、Helotiales(柔膜菌目)占比0.0816%、Microascales占比0.1617%、Russulales占比0.1415%、Sordariomycetes占比0.0930%、Trichosporonales占比0.1424%、Wallemiales占比0.0855%。在属的分类水平上将真菌群落分为优势类群、常见类群、次常见类群和稀有类群四大类。优势类群为比例超过10%的真菌群落,包括Cryptococcus(隐球菌属)占比31.57%、Alternaria(链格孢属)占比18.21%、Pleospora(格孢腔菌属)占比10.90%;常见类群为比例小于10%的大于1%的真菌群落,包括Fungiunclassified占比8.58%、Davidiella占比4.86%、Epicoccum(附球菌属)占比2.94%、Cladosporium(枝孢属)占比2.4023%、Cystofilobasidium占比2.2694%、Fusarium(镰刀菌属)占比1.6913%、Gibberella(赤霉菌属)占比1.5467%、Incertaesedis占比1.3483%、Udeniomyces占比1.2964%、Pyrenophora(核腔菌属)占比1.0122%、Tremellomycetes占比4.6574%。次常见类群指比例大于0.5%,小于1%的真菌类群:包括Aspergillus占比0.6490%,Dothioraceae占比0.7199%,Phoma占比0.7334%,Pleosporales占比0.5809%;比例小于0.5%,大于0.1%的稀有类群,包括Ascomycota占比0.2396%,Basidiomycota占比0.1317%,Blumeria占比0.2186%,Dioszegia占比0.1690%,Hannaella占比0.2735%,Lactarius占比0.1157%,Pseudallescheria占比0.1202%,Stagonospora占比0.1183%,Trichosporon占比0.1424%,Sporidiobolales占比0.1855%,Sporidiobolus占比0.4722%,Sporobolomyces占比0.2884%。另外,还有占比小于0.1%的超稀有类群151种,包括Incertae占比0.0930%,Leptosphaeria占比0.0115%,Microascus占比0.0359%,Mortierella占比0.0247%,Mycosphaerellaceae占比0.0227%,Trichothecium占比0.0185%,Wallemia占比0.0855%,Nectriaceae占比0.0580%,Penicillium占比0.0726%,Phaeosphaeriaceae占比0.0821%,Pleosporaceae占比0.0701%,Russula占比0.0258%,Sclerotiniaceae占比0.0717%,Setosphaeria占比0.0647%,Sordariomycetes占比0.0930%。(6)小麦种子与筛下物进行比较的结果在属的分类水平上,可以看出澳大利亚小麦种子与筛下物病原真菌类群差异较大,种子优势类群依次为Alternaria,Pleospora,Cryptococcus,Fungi_unclassified,Tremellomycetes,Fusarium,Cladosporium,Epicoccum,Udeniomyces,Dothioraceae,Pyrenophora,Davidiella。筛下物优势类群依次为Cryptococcus,Alternaria,Davidiella,Tremellomycetes_unclassified,Epicoccum,Cystofilobasidium,Gibberella,Pleospora,Phoma。详细结果如表2所示。表2属的水平上澳大利亚小麦和筛下物携带真菌OTU数目从表2可以看出种子携带病原真菌占比最高的依次是Alternaria(链格孢属)占比21.38%,Pleospora占比20.40%,Cryptococcus(隐球菌属)占比19.12%。筛下物携带病原真菌占比最高的依次是Cryptococcus(隐球菌属)占比44.02%,Alternaria(链格孢属)占比15.0384%,Davidiella占比8.6786%。可见种子和筛下物所携带病原真菌种类和比例是有差异的。本例进一步在属的水平上将AW1与AW2数据整合,AW3与AW4数据整合,AW5与AW6数据整合,AW7与AW8数据整合。也就是将2015年5月、7月、10月、11月四批样品包含种子与筛下物进行属水平分类。结果显示,四批样品的整合结果,与8份样品的整合结果一致,优势菌群依次为:Cryptococcus(隐球菌属)、Alternaria(链格孢属)、Pleospora(格孢腔菌属);常见类群为比例小于10%的大于1%的真菌群落,包括Fungiunclassified占比8.58%、Davidiella占比4.86%、Epicoccum(附球菌属)占比2.94%、Cladosporium(枝孢属)占比2.4023%、Cystofilobasidium占比2.2694%、Fusarium(镰刀菌属)占比1.6913%、Gibberella(赤霉菌属)占比1.5467%、Incertaesedis占比1.3483%、Udeniomyces占比1.2964%、Pyrenophora(核腔菌属)占比1.0122%。综上所述,本例的所有小麦种子和筛下物样品中,通过高通量测序及分析得到可分类OTU共622个,分属于5个门:Ascomycota、Basidiomycota、Chytridiomycota、Fungi_unclassified、Zygomycota;64个目,其中优势类群为Tremellales(银耳目)、Pleosporales(假球壳目)、Capnodiales、Cystofilobasidiale、Fungi_unclassified、Hypocreales、Tremellomycetes、Trichosphaeriales;201个属,其中优势类群为Cryptococcus(隐球菌属)、Alternaria(链格孢属)、Pleospora(格孢腔菌属)、Fungi_unclassified、Davidiella、Epicoccum(附球菌属)、Cladosporium(枝孢属)、Cystofilobasidium、Fusarium(镰刀菌属)、Gibberella(赤霉菌属)、Incertaesedis、Udeniomyces、Pyrenophora(核腔菌属)、Tremellomycetes。并且,不同样品间OTU种类数目各不相同:样品AW1为155,样品AW2为281,样品AW3为119,样品AW4为108,样品AW5为81,样品AW6为201,样品AW7为122,AW8为148。可见筛下物的OTU数量是大于种子的OUT数量。并且种子与筛下物携带的病原真菌的种类和占比有小幅差异,种子携带的病原真菌比例最高依次为Alternaria(链格孢属),Pleospora,Cryptococcus(隐球菌属),这三个属占筛下物携带病原真菌总量的60%。筛下物携带的病原真菌依次是Cryptococcus(隐球菌属),Alternaria(链格孢属),Davidiella,这三个属占筛下物携带病原真菌总量的70%。通过高通量测序测得澳大利亚小麦种子携带的病原真菌共124个属,筛下物携带的病原真菌共167个种属,种子和筛下物都携带的病原真菌96个属。可见筛下物携带的病原真菌数量和种类远多于种子携带的病原真菌。通过高通量测序技术对进境澳大利亚小麦上病原真菌的检测可以看出高通量测序技术克服了传统分离培养方法中有些病原物专性寄生、培养条件苛刻、在群体中丰度低、微生物存活但不能培养的制约,克服了梯度凝胶电泳DGGE和TGGE、末端限制性片段长度多态性分析T-RFLP等基于PCR的分子技术耗时长、通量低、信息量小、成本高等缺陷,可以直接对进境小麦种子或者筛下物进行DNA的提取、文库的构建、上机测序、数据分析,从而得到样品中病原微生物的信息,简化了研究步骤,缩短了研究周期,相对于过去方法能检测到更多的基因和未知的转录组并且能检测到表达丰度很低的基因,具有更好的准确性、灵敏性,为口岸通关节约的时间成本。以上内容是结合具体的实施方式对本申请所作的进一步详细说明,不能认定本申请的具体实施只局限于这些说明。对于本申请所属
技术领域:
的普通技术人员来说,在不脱离本申请构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本申请的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种构建Hi‑C高通量测序文库的方法与制造工艺
- 桑葚病原菌高通量鉴定及种属分类方法及其应用与制造工艺
- 基于高通量测序技术检测乳腺癌/卵巢癌易感基因的多重PCR特异性引物、试剂盒及方法与制造工艺
- 基于高通量测序检测人EGFR基因变异的质控方法及试剂盒与制造工艺
- 基于高通量测序技术的小麦病原微生物检测方法及其应用与制造工艺
- 一种基于二代高通量测序技术的地中海贫血症的基因检测试剂盒的制造方法与工艺
- 一种基于集群的高通量数据分析方法与制造工艺
- 高通量双折射干涉成像光谱仪装置及其成像方法与制造工艺
- 高通量测序检测人循环肿瘤DNA EGFR基因的捕获探针及试剂盒的制造方法与工艺
- 一种高通量测序检测无创亲子鉴定的方法与制造工艺