R,S‑告依春的制备方法与流程
本发明涉及一种中药有效成分的提取分离工艺领域,具体的,本发明涉及一种R,S-告依春的制备方法。
背景技术:
R,S-告依春是板蓝根中的一种有效活性成分,具有显著的抗病毒活性。传统的制备R,S-告依春的方法基本采用大孔树脂柱分离,大多使用大孔树脂柱分离层析、正相柱层析及反相柱层析联用的方法,然而采用这种方法制备出的样品得率低、制备量小、纯度不高,无法进行规模化的制备。此外,这种方法还存在污染大,耗时长,成本高,且在制备过程中因使用到大量的吸附填料,使样品原有的生物活性大打折扣,所得R,S-告依春活性降低,生产效益低下,不适合推广使用。
技术实现要素:
基于此,本发明在于克服现有技术的缺陷,提供一种用高速逆流大量制备R,S-告依春的方法,利用所述方法制备R,S-告依春质量高,单次制备量大,并对样品活性无影响,也无外加污染,可用于药品,化妆品,保健品等。
其技术方案如下:
一种制备R,S-告依春的方法,包括如下步骤:
S1.提取:取板蓝根药材为原料,蒸馏水超声提取,溶剂萃取后浓缩得到主含R,S-告依春的提取物;
S2.高速逆流色谱分离:配置体积比为7~9:0.5~1.5:0.5~1.5:8~10的乙酸乙酯-正丁醇-乙醇-水的溶剂体系,所述溶剂体系包括作为固定相的上层和作为流动相的下层,取适量所述流动相溶解所述提取物,将所述溶剂体系泵入高速逆流色谱的色谱柱中,然后将溶解后的所述提取物泵入高速逆流色谱洗脱分离,并根据记录仪得到的逆流色谱图谱判定流份中是否含有R,S-告依春,收集含有R,S-告依春的流份,干燥得到R,S-告依春粗品,其中,所述高速逆流色谱的参数设置如下:转速为300~500rpm,分离温度为17~23℃,检测波长为240~260nm,流动相的流速为9~20ml/min;
S3.正相柱层析分离:以正相层析硅胶为填料、体积比为5~9:1~3的石油醚-乙酸乙酯为洗脱溶剂进行正相柱层析所述分离R,S-告依春粗品,洗脱体积为15~25倍柱体积,所述R,S-告依春粗品与填料的质量比为1:20~1:50,收集含有R,S-告依春的洗脱液,减压蒸馏即得。
本发明采用高速逆流色谱分离R,S-告依春,由于高速逆流色谱不需要固体支撑体,R,S-告依春的分离仅依据其在固定相、流动相中分配系数的不同就可实现,无需大量使用吸附填料,因而避免了因不可逆吸附而引起的样品损失、失活、变性等,不仅使R,S-告依春能够充分回收,回收的R,S-告依春更能保持其本来的特性,且由于被分离的R,S-告依春提取物与液态固定相之间能够充分接触,使得样品的制备量大大提高。
在其中一个实施例中,步骤S2所述乙酸乙酯-正丁醇-乙醇-水的混合溶液中乙酸乙酯-正丁醇-乙醇-水的体积比为9:1:1:9。
在其中一个实施例中,步骤S2中所述固定相的保留值为80~90%。
在其中一个实施例中,步骤S2中所述固定相的保留值为82.4%。
在其中一个实施例中,步骤S2中所述流动相的流速为10~15ml/min。
在其中一个实施例中,步骤S2中所述转速为400~500rpm。
在其中一个实施例中,步骤S2中所述检测波长为254nm。
在其中一个实施例中,所述步骤S1为:取板蓝根药材为原料粉碎过筛,蒸馏水超声提取,过滤,取滤液调碱后采用乙酸乙酯萃取,取乙酸乙酯层,浓缩除去乙酸乙酯得到主含R,S-告依春的提取物。
在其中一个实施例中,所述步骤S1为:取板蓝根药材为原料,粉碎过筛,按料液比1:3~1:10用蒸馏水溶解,30~70W的功率下超声30~60min,提取3~5次,过滤,取滤液,用氢氧化钠将滤液调pH为8~12,采用乙酸乙酯萃取,取乙酸乙酯层,浓缩除去乙酸乙酯得到主含R,S-告依春的提取物。
在其中一个实施例中,步骤S3所述洗脱溶剂中石油醚和乙酸乙酯的体积比为8:2。
在其中一个实施例中,步骤S3中所述R,S-告依春粗品与填料的质量比为1:25。
在其中一个实施例中,所述步骤S3中采用正相薄层色谱判定洗脱液中是否含有R,S-告依春。
在其中一个实施例中,步骤S2中所述混合溶液泵入高速逆流色谱的色谱柱的过程为:混合溶液的上层液于仪器静态泵入高速逆流色谱柱中,混合溶液的下层液于仪器动态泵入色谱柱中,上相为固定相,下相为流动相。
在其中一个实施例中,步骤S2中所述溶解后的提取物在固定相、流动相平衡后被泵入色谱柱中。
在其中一个实施例中,所述高速逆流色谱柱的体积为1000ml。
在其中一个具体实施例中,单次通过单根高速逆流色谱柱的提取物质量为3~5g。
在其中一个实施例中,所述硅胶为100~200目硅胶。
在其中一个实施例中,步骤S3中R,S-告依春粗品经洗脱溶剂溶解后再进行正相柱层析分离。
在其中一个实施例中,步骤S3中减压蒸馏制备目标物的过程中同时回收溶剂。
本发明的有益效果在于:
本发明采用高速逆流色谱仪提纯分离制备R,S-告依春,再经正相柱层析制备得到高纯的R,S-告依春,该制备方法耗时短,R,S-告依春质量高,单次制备量大,可进行规模化的制备且成本低;该制备方法因未大量使用吸附填料,对样品活性影响较小,保留了原有的生物活性;制备出的产品也无外加污染,可用于药品,化妆品,保健品等。
附图说明
图1为制备R,S-告依春的工艺流程图。
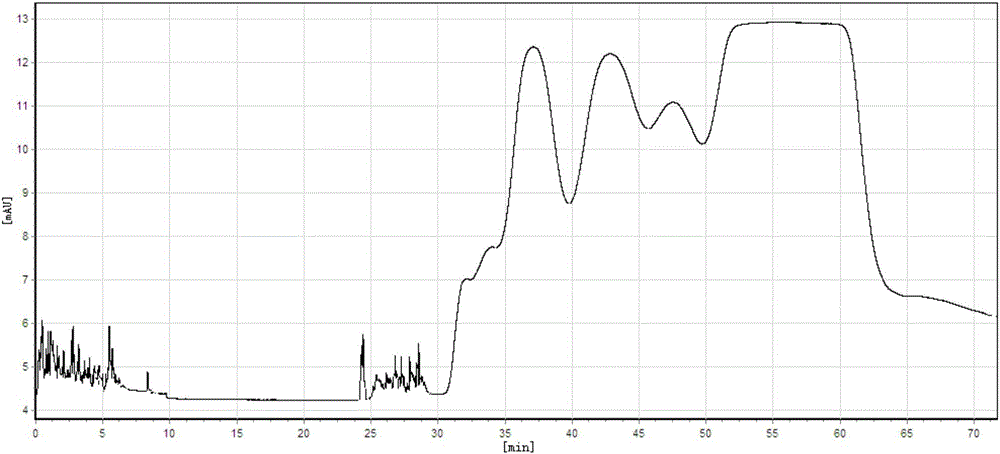
图2为实施例1中记录仪记录的高速逆流色谱分离R,S-告依春的逆流色谱图谱。
具体实施方式
为使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及具体实施方式,对本发明进行进一步的详细说明。应当理解的是,此处所描述的具体实施方式仅用以解释本发明,并不限定本发明的保护范围。
图1为制备R,S-告依春的工艺流程图。以下各实施例所用原料均为普通市售产品,正相层析硅胶的目数为100~200目。
实施例1
按下列步骤制备告依春:
S1.提取:取板蓝根药材13.3千克为原料,粉碎过筛,按料液比1:6用蒸馏水溶解,60W的功率下超声提取60min,每隔20分钟搅拌,每超声提取一次后静置60分钟,期间每隔30分钟搅拌一次,如此操作提取4次,过滤,取滤液,另置,滤渣按上述操作反复提取3次,合并滤液。用氢氧化钠将水提液调pH为10时,采用乙酸乙酯萃取,萃取3次,合并乙酸乙酯层,低压浓缩除去乙酸乙酯得到主含R,S-告依春的提取物27.21克。
S2.高速逆流色谱分离:配置大量体积比为9:1:1:9的乙酸乙酯-正丁醇-乙醇-水的溶剂体系,所述溶剂体系包括作为固定相的上层和作为流动相的下层,取280mL流动相溶解上述提取物,将溶剂体系的上层液于仪器静态泵入高速逆流色谱柱中,溶剂体系的下层液于仪器动态泵入色谱柱中,溶解后的提取物在固定相、流动相平衡后被泵入色谱柱中,经高速逆流色谱洗脱分离,接收流份,并根据记录仪得到的逆流色谱图谱判定流份中是否含有目标产物,收集含有目标产物的流份,干燥得到R,S-告依春粗品,其中,所述高速逆流色谱的参数设置如下:转速为400rpm,分离温度为17~23℃,检测波长为254nm,流动相的流速为10ml/min,固定相保留值为82.4%;
S3.正相柱层析分离:以正相层析硅胶为填料、体积比为8:2的石油醚-乙酸乙酯为洗脱溶剂进行正相柱层析分离R,S-告依春粗品,所述R,S-告依春粗品层析前用洗脱溶剂溶解。洗脱体积为15~25倍柱体积,根据正相薄层色谱判定洗脱液中是否含有R,S-告依春,收集含有R,S-告依春的洗脱液(第11~22柱体积),减压蒸馏得目标物9.5克,其中,所述R,S-告依春粗品与填料的质量比为1:25。
图2为实施例1中记录仪记录的高速逆流色谱分离R,S-告依春的逆流色谱图谱,当观察到出现此峰型的图谱时表明流份中含有R,S-告依春。
所得R,S-告依春经HPLC检测纯度为99.9%。
所得R,S-告依春粗品的核磁数据:6.01(ddd,J=17.1,10.3,6.7,1H),5.48(d,J=17.1,1H),5.37(d,J=10.5,2H),3.91(t,J=9.6,1H),3.48(dd,J=10.1,7.6,1H)。
R,S-告依春化学结构式如式1所示:
实施例2
按下列步骤制备告依春:
S1.提取:与实施例1中的步骤S1相同;
S2.高速逆流色谱分离:与实施例1中的步骤S2相似,区别在于乙酸乙酯-正丁醇-乙醇-水的体积比为7:1.5:0.5:8,固定相保留值为81.3%;
S3.正相柱层析分离:与实施例1中的步骤S3相同,得到R,S-告依春9.48g。
所得R,S-告依春经HPLC检测纯度为99.7%。
实施例3
按下列步骤制备告依春:
S1.提取:与实施例1中的步骤S1相同;
S2.高速逆流色谱分离:与实施例1中的步骤S2相似,区别在于乙酸乙酯-正丁醇-乙醇-水的体积比为9:0.5:1.5:10,固定相保留值为83.7%;
S3.正相柱层析分离:与实施例1中的步骤S3相同,得到R,S-告依春9.53g。
所得R,S-告依春经HPLC检测纯度为99.3%。
实施例4
按下列步骤制备告依春:
S1.提取:与实施例1中的步骤S1相同;
S2.高速逆流色谱分离:与实施例1中的步骤S2相似,区别在于转速为500rpm;
S3.正相柱层析分离:与实施例1中的步骤S3相同,得到R,S-告依春9.47g。
所得R,S-告依春经HPLC检测纯度为99.5%。
实施例5
按下列步骤制备告依春:
S1.提取:与实施例1中的步骤S1相同;
S2.高速逆流色谱分离:与实施例1中的步骤S2相似,区别在于转速为300rpm;
S3.正相柱层析分离:与实施例1中的步骤S3相同,得到R,S-告依春9.54g;
所得R,S-告依春经HPLC检测纯度为99.1%。
实施例6
按下列步骤制备告依春:
S1.提取:与实施例1中的步骤S1相同;
S2.高速逆流色谱分离:与实施例1中的步骤S2相似,区别在于流动相的流速为15ml/min;
S3.正相柱层析分离:与实施例1中的步骤S3相同,得到R,S-告依春9.5g。
所得R,S-告依春经HPLC检测纯度为99.8%。
实施例7
按下列步骤制备告依春:
S1.提取:与实施例1中的步骤S1相同;
S2.高速逆流色谱分离:与实施例1中的步骤S2相似,区别在于流动相的流速为20ml/min;
S3.正相柱层析分离:与实施例1中的步骤S3相同,得到R,S-告依春9.52g。
所得R,S-告依春经HPLC检测纯度为99.3%。
实施例8
按下列步骤制备告依春:
S1.提取:与实施例1中的步骤S1相同;
S2.高速逆流色谱分离:与实施例1中的步骤S2相似,区别在于流动相的流速为9ml/min;
S3.正相柱层析分离:与实施例1中的步骤S3相同,得到R,S-告依春9.54g。
所得R,S-告依春经HPLC检测纯度为99.1%。
实施例9
按下列步骤制备告依春:
S1.提取:与实施例1中的步骤S1相同;
S2.高速逆流色谱分离:与实施例1中的步骤S2相同;
S3.正相柱层析分离:与实施例1中的步骤S3相似,区别在于石油醚-乙酸乙酯的体积比为5:3,得到R,S-告依春9.53g;
所得R,S-告依春经HPLC检测纯度为99.3%。
实施例10
按下列步骤制备告依春:
S1.提取:与实施例1中的步骤S1相同;
S2.高速逆流色谱分离:与实施例1中的步骤S2相同;
S3.正相柱层析分离:与实施例1中的步骤S3相似,区别在于石油醚-乙酸乙酯的体积比为9:1,得到R,S-告依春9.52g。
所得R,S-告依春经HPLC检测纯度为99.4%。
实施例11
按下列步骤制备告依春:
S1.提取:与实施例1中的步骤S1相同;
S2.高速逆流色谱分离:与实施例1中的步骤S2相同;
S3.正相柱层析分离:与实施例1中的步骤S3相似,区别在于R,S-告依春粗品与填料的质量比为1:1,得到R,S-告依春9.56g;
所得R,S-告依春经HPLC检测纯度为99.0%。
实施例12
按下列步骤制备告依春:
S1.提取:与实施例1中的步骤S1相同;
S2.高速逆流色谱分离:与实施例1中的步骤S2相同;
S3.正相柱层析分离:与实施例1中的步骤S3相似,区别在于R,S-告依春粗品与填料的质量比为1:50,得到R,S-告依春9.53g;
所得R,S-告依春经HPLC检测纯度为99.4%。
对比例1
按下列步骤制备告依春:
S1.提取:与实施例1中的步骤S1相同;
S2.高速逆流色谱分离:与实施例1中的步骤S2相似,区别在于乙酸乙酯-正丁醇-乙醇-水的体积比为10:1:2:9,固定相保留值为71.2%;
S3.正相柱层析分离:与实施例1中的步骤S3相同,得到R,S-告依春为9.9g。
所得R,S-告依春经HPLC检测纯度为89.1%。
对比例2
按下列步骤制备告依春:
S1.提取:与实施例1中的步骤S1相同;
S2.高速逆流色谱分离:与实施例1中的步骤S2相同,得到R,S-告依春9.7g。
所得R,S-告依春经HPLC检测纯度为97.5%。
从实施例1至12可知,利用本发明所述方法制备的R,S-告依春的纯度高达99%且单次产量高,此外,由于该制备过程仅通过高速逆流色谱与正相层析柱,比传统工艺更加简便和省时。通过实施例1和对比例1的对比可知,步骤S2中乙酸乙酯-正丁醇-乙醇-水的用量比例对R,S-告依春的纯度影响较大,超过一定范围,其纯度会显著下降。通过实施例1和对比例2的对比可知,步骤S3对R,S-告依春的纯度影响也较大,正相柱层析可进一步提高R,S-告依春的纯度。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!