一种16‑羟基‑9‑烯‑十六羧酸酯的合成方法与流程
本发明涉及有机化工领域,特别涉及一种16-羟基-9-烯-十六羧酸酯的合成方法。
背景技术:
对于自然界存在的香料化合物,其中最重要的一个类别就是具有烯烃结构的大环内类烯烃化合物。该类型化合物均具有优雅柔和的香型,定香性能良好,接近天然香料的典型气味。由于直接从天然获取该类型的化合物,成本比较高,并且对自然环境造成巨大损害。因此研究其简单高效的合成方法成为发展的必然。由于该类型的化合物主要结构为大环酮类和内酯类化合物烯烃化合物,其合成的关键在于烯烃的生成与环化。对于烯烃化反应,经常采用邻二醇的化合物经过脱水烯化而来,而对于分子内的成环化反应,根据原料结构的不同,其成环方式也有所区别,从成环的方式而言,主要包括了以酰氧键碳氧键及碳碳键等成环方式。对于酮式大环类化合物,通常可采用羟醛缩合法来合成制备,反应条件温和,目前该类化合物的生产已得到较好解决。
而大环内酯烯化合物仍存在较大的合成难度,其已经作为合成大环麝香香料的研究热点之一。大环内酯的基本结构是12元环以上且环骨架上含有一个或多个酯基的分子化合物,传统的合成路线常使用卤代长链烷酸作为原料进行生产,但原料加工路线长,卤化物的添加导致环境污染严重(2000,TL,41(45):8673)。
该方法先用15%的溴化氢乙酸溶液溴化,酯化,烯烃化,脱保护得到关键中间烯基羧酸类化合物,该化合物进一步环合或者即可制备酯化或酮基化产物。其溴化氢的过量使用,对生成设备和环境造成了极大的污染,因此,使得人们逐渐将注意力转移到绿色环保的合成方法研究的研究上来。
由于合成该类型化合物多采用多羟基脂肪羧酸作为起始原料,其分子中含有一个游离的羧基和三个游离的羟基,其本身能够作为合成大环内酯的一种原料,在很大程度上减少了繁琐冗余有毒有害的中间合成过程,从而降低合成成本。但由于分子结构中端位的碳原子所连接的羧基及羟基基团,导致其成环的过程是以形成酰氧键的方式进行的,即活化分子中一端的羧基,使之与另一端的羟基反应生成大环内酯的方法。其直接的内酯化过程,除了存在分子间酯化连接外,其分子内产物亦存在如下三种可能的结构,其结构具有有一定的相似性,分离困难。
为了避免生成上述混合物,人们往往采用分步的方法进行合成,其已有合成方法主要可以分为两种类型,第一种,主要采用选择性的保护基,现对领位二羟基进行保护,然后端基环取代环化,再进行烯烃化获取目标物质;第二种,类似于卤代合成类似的方法,先选择合适的保护基团对碳链端头的羟基和羧基进行保护,烯化,脱保护和环化得到目标产物。
第一种方法,常采用邻二醇的缩醛保护,避免9和10位的内酯化的基础上,再经过脱叉烯化,内酯化得到目标品,或者先内酯化,然后脱叉烯化反应得到目标产物。Shiina等在樟脑磺酸(CSA)的催化下(T,2006,62(33):7934;TL,2002,43(42):7535;TL,2010,51(17):2348;TL,1999,40(6):1123),紫胶酮酸与PhCH(OMe)2发生缩醛反应得苄叉保护中间产物,经活性酯——2-甲基-6-硝基苯甲酸酐(MNBA)和4-二甲氨基吡啶氧化物(DMAPO)催化发生内酯化反应获得单体内酯中间产物,在醋酸催化下水解叉苄叉,其所得邻二醇与硫代羰基二咪唑反应形成硫代碳酸酯,然后还原脱硫形成卡宾,分子内电子转移脱掉一分子二氧化碳形成双键的目标产物,其反应过程如下;在这个过程亦有研究者直接利用N,N-二甲基甲酰胺二新戊基乙缩醛(DMF-DEA)进行叉化保护。然后在酸酐的作用下,直接脱叉烯烃化得到目标产物,减少了反应步骤。
另外一种是利用具有大位阻效应的活性酯催化剂,选择性的端基内酯化,然后再脱邻二醇成烯烃化反应,如Venkataraman等(TL,1980,21(19):1893;TL,1979,20(32):3037)利用三聚氯氰(TCT)催化长链-羟基酸进行内酯化反应,在丙酮中先后加入三聚氯氰(TCT)和三胺(Et3N),混合于室温条件下反应得到产物,如合成(9E)-异黄葵内酯的中间产物。其最终产物收率高达86%。同理由于N,N-二甲基甲酰胺二新戊基乙缩醛(DMF-DEA)对高位阻化合物能够显示出独特的优越性,且能与很多含有活泼氢的化合物反应,已经越来越被人们用于醚硫醚等杂环化合物的合成中。Villemin在氮气保护下(Synthesis,1987(2):154;TL,2006,47(31):5553),就利用DMF-DEA在甲苯混合,发生亲电取代生成,然后经醋酸酐脱水减压蒸馏收集得到目标产物。
以前所有制备方法均需要分步进行,反应步骤较长,生产成本高;且中间产物还需经过硅胶柱层析分离纯化才可以得到适合下一步反应的原料,不利于大规模生产使用。
技术实现要素:
本发明的发明目的是提供一种16-羟基-9-烯-十六羧酸酯的合成方法。该方法中多羟基脂肪羧酸经过一锅法进行端羟基和羧酸的保护,以及领羟基的烯烃活化,不经任何分离操作,加入催化剂直接将领羟基脱水烯烃化,反应结束直接加水分解保护基,即得到目标产物。且该方法操作简单,反应所涉及到试剂对环境友好,目标产物分离纯化简单。
为了实现以上目的,本发明的技术方案为:
一种16-羟基-9-烯-十六羧酸酯的合成方法,该方法包括以下步骤:
(1)、采用多羟基脂肪酸作为反应起始原料,如9,10,16-三羟基十六羧酸(1.0eq);
(2)、选择的保护试剂为原酸缩酸酯类化合物,如原甲酸三甲酯,原甲酸三乙酯或乙酸三甲酯等,其与原料的配比为2.5~10eq;
(3)、合成反应温度为50~120℃,反应时间为2~8h;
(4)、烯烃化反应催化剂为各种类型的酸酐类催化剂,如乙酸酐,丙酸酐,丁酸酐和苯甲酸酐,其与原料的配比为1.0~2.0eq;
烯烃化反应温度为110~180℃,反应时间为3~8h。
该方法具体包括以下步骤:
取1当量(1.0eq)9,10,16-三羟基十六羧酸加入到单口反应瓶中,然后加入3倍体积的硅油和2.5~10eq的原酸缩酸酯(原甲酸三甲酯),氮气保护下,控温在50~120℃反应2~8h;然后加入约1.0~2.0eq的酸酐,控温在110~180℃反应约3~8h。冷却到室温,加入一定量的H2O,搅拌水解约30min,然后调节pH到5.0,乙酸乙酯萃取,无水硫酸钠干燥,旋转蒸干得到粗品16-羟基-9-烯-十六羧酸酯。
精制的方法包括以下步骤:
上述产品溶解在一定量的水中,用氢氧化钠调节pH到9.0~9.5,样品溶解完全,用石油醚萃取三次,然后水层调节pH到5.0,乙酸乙酯萃取,合并萃取液,无水硫酸钠干燥,过滤,旋转蒸干。固体中加入约一定量的无水乙醇重结晶获得目标产物,NMR分析结构正确。
本发明的有益效果是:
(一)、本研究根据端基羟基和羧基位阻比较小的特点,开发出了一锅法合成的该类型化合物关键中间体——16-羟基-9-烯-十六羧酸酯的合成方法。
(二)、其多羟基脂肪羧酸经过一锅法进行端羟基和羧酸的保护,以及领羟基的烯烃活化,不经任何分离操作,加入催化剂直接将领羟基脱水烯烃化,反应结束直接加水分解保护基,即得到目标产物。
(三)、该方法操作简单,反应所涉及到试剂对环境友好,目标产物分离纯化简单。
(四)、分离纯化简单,有利于大规模工业化生产。
附图说明
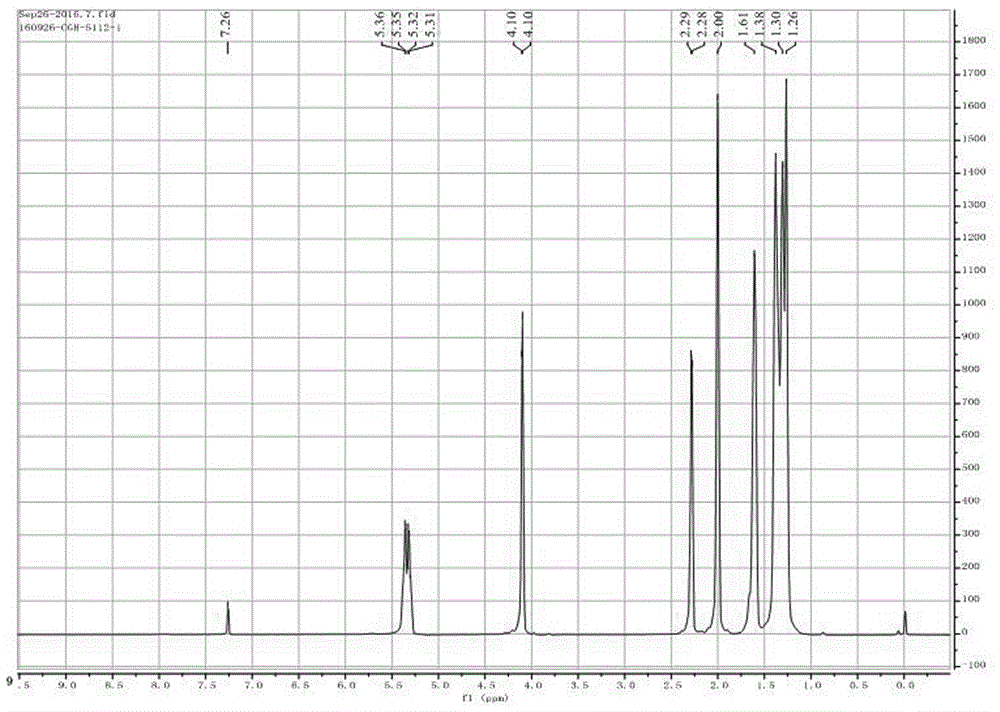
图1为9-烯-十六羧酸内酯H1NMR图
图2为9-烯-十六羧酸内酯C13NMR图
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明的内容之后,本领域技术人员可以对本发明作各种改动或修改,但这些等价形式同样落于本申请所附权利要求书所限定的范围。
实施例1:
取1.0g(3.3mmol)9,10,16-三羟基十六羧酸加入到单口反应瓶中,然后加入约3.0mL的硅油和1.0mL(3.0eq)的原甲酸三乙酯,氮气保护下,控温在50℃反应2h,然后升温到120℃反应6h;冷却低于50℃,迅速一次性的加入0.31mL(1.0eq)的乙酸酐,控温在110℃反应h,然后升温到180℃反应约7h。冷却到室温,加入30mL的H2O,搅拌水解约30min,然后调节pH到5.0,用乙酸乙酯萃取(20mL×3),无水硫酸钠干燥,旋转蒸干得到粗品16-羟基-9-烯-十六羧酸酯。
实施例2:
取1.0g(3.3mmol)9,10,16-三羟基十六羧酸加入到单口反应瓶中,然后加入约3.0mL的硅油和2.0mL(6.0eq)的原甲酸三乙酯,氮气保护下,控温在60℃反应4h,然后升温到120℃反应6h;冷却低于50℃,迅速一次性的加入0.47mL(1.5eq)的乙酸酐,控温在110℃反应2h,然后升温到180℃反应约7h。冷却到室温,加入30mL的H2O,搅拌水解约30min,然后调节pH到5.0,用乙酸乙酯萃取(20mL×3),无水硫酸钠干燥,旋转蒸干得到粗品16-羟基-9-烯-十六羧酸酯。
实施例3:
取1.0g(3.3mmol)9,10,16-三羟基十六羧酸加入到单口反应瓶中,然后加入约3.0mL的硅油和3.0mL(9.0eq)的原甲酸三乙酯,氮气保护下,控温在70℃反应3h,然后升温到120℃反应6h;冷却低于50℃,迅速一次性的加入0.56mL(1.8eq)的乙酸酐,控温在110℃反应1h,然后升温到180℃反应约7h。冷却到室温,加入30mL的H2O,搅拌水解约30min,然后调节pH到5.0,用乙酸乙酯萃取(20mL×3),无水硫酸钠干燥,旋转蒸干得到粗品16-羟基-9-烯-十六羧酸酯。
实施例4:
取1.0g(3.3mmol)9,10,16-三羟基十六羧酸加入到单口反应瓶中,然后加入约3.0mL的硅油和4.0mL(10.0eq)的原甲酸三乙酯,氮气保护下,控温在80℃反应2h,然后升温120℃反应6h;冷却低于50℃,迅速一次性的加入0.62mL(2.0eq)的乙酸酐,控温在110℃反应1h,然后升温到180℃反应约7h。冷却到室温,加入30mL的H2O,搅拌水解约30min,然后调节pH到5.0,用乙酸乙酯萃取(20mL×3),无水硫酸钠干燥,旋转蒸干得到粗品16-羟基-9-烯-十六羧酸酯。
精制:
取实施例1中获取的0.6g产品16-羟基-9-烯-十六羧酸酯溶解在约30mL的水中,用5wt%的氢氧化钠溶液调节pH到9.0~9.5,样品溶解完全,用石油醚萃取3次,每次20ml,即(20mL×3),然后水层用1.0M的盐酸调节pH到5.0,乙酸乙酯萃取(20mL×3),合并萃取液,无水硫酸钠干燥,过滤,旋转蒸干。固体中加入约15mL的无水乙醇重结晶获得目标产物约0.67g,收率为75%。
验证案例:
上述精制的16-羟基-9-烯-十六羧酸酯约1.0g加入到单口瓶中,加入20mL的无水甲苯和0.2g的对甲苯磺酸,加热回流分水反应约4h,冷却到室温,加入约20mL的H2O洗涤,调节水层pH到中性(20mL×2),无水硫酸钠干燥,过滤,旋转蒸干得到无色油状液体,NMR分析目标产物9-烯十六环内酯(13C NMR(151MHz,Chloroform-d)δ174.20,131.33,130.75,64.52,35.06,31.94,31.57,29.79,29.05,28.73,28.14,28.09,28.02,27.10,26.94,25.19.1H NMR(600MHz,Chloroform-d)δ5.34(dd,J=21.9,6.8Hz,2H),4.10(d,J=4.5Hz,2H),2.28(d,J=7.0Hz,3H),2.00(s,4H),1.61(s,3H),1.49–1.12(m,14H))。
以上所述实例仅是本专利的优选实施方式,但本专利的保护范围并不局限于此。应当指出,对于本技术领域的普通技术人员来说,在不脱离本专利原理的前提下,根据本专利的技术方案及其专利构思,还可以做出若干改进和润饰,这些改进和润饰也应视为本专利的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!