一种基于氧化吡啶配体的金属有机框架及制备方法与应用与流程
本发明属于催化剂制备技术领域,具体涉及一种基于氧化吡啶配体的金属有机框架及制备方法与应用。
背景技术:
醛或酮与活性亚甲基化合物的脱水缩合反应,形成碳碳双键的反应称为Knoevenagel缩合反应。此类反应能够直接合成大量有用的化合物,在工业、农业、药学、生物科学等诸多领域有着广泛应用。Knoevenagel缩合反应一般是用Lewis酸或碱为催化剂,亦可采用弱碱(胺、吡啶等)作催化剂,在均相或异相环境中进行反应,一般所需时间较长,而且产率较低。
金属-有机框架材料(Metal-OrganicFrameworks,MOFs)是一类有机-无机杂化材料,由有机配体和无机金属单元构建而成。一般具有多变的拓扑结构以及物理化学性质。与传统的多孔材料相比,MOFs具有较大的比表面积和框架内孔体积,作为一类功能化材料在催化方面表现出很大的优势。
然而现有金属有机框架作为一种路易斯酸催化剂存在的问题有稳定性差、制备步骤繁琐、活性低、不能回收等。
技术实现要素:
为克服现有技术的缺陷,本发明提供了一种基于氧化吡啶配体的金属有机框架及制备方法与应用。
为实现上述目的,本发明的技术方案为:
一种氧化吡啶配体L,其中,L的化学结构式为:
上述氧化吡啶配体L的制备方法,(1)将3,5-二卤代吡啶制备成3,5-二卤代吡啶-N-氧化物,(2)将3,5-二卤代吡啶-N-氧化物经过Suzuki偶联反应获得氧化吡啶配体L。
所述Suzuki偶联反应为也称作铃木反应,是一个较新的有机偶联反应,零价钯配合物催化下,芳基、吡啶或烯基硼酸(硼酸酯)与氯、溴、碘代芳烃(吡啶或烯烃)发生交叉偶联。
3,5-二卤代吡啶的结构式为:
其中,R为卤代基团,如Cl、Br等。
3,5-二卤代吡啶-N-氧化物的结构式为:
其中,R为卤代基团,如Cl、Br等。
优选的,(1)采用间氯过氧苯甲酸将3,5-二溴吡啶制备成3,5-二溴吡啶-N-氧化物,(2)将3,5-二溴吡啶-N-氧化物与4-吡啶硼酸经过Suzuki偶联反应获得氧化吡啶配体L。
优选的,步骤(1)采用的溶剂为四氢呋喃。
优选的,步骤(1)中的反应条件为:在常温下搅拌48h。
进一步优选的,步骤(1)中3,5-二溴吡啶与间氯过氧苯甲酸的摩尔比为1:2.50~3.00。
更进一步优选的,步骤(1)中3,5-二溴吡啶与间氯过氧苯甲酸的摩尔比为1:2.96。
优选的,步骤(2)采用的催化剂为四(三苯基膦)钯。
优选的,步骤(2)的反应条件为:加热回流48小时,加热回流温度为75-80℃。
优选的,步骤(2)中3,5-二溴吡啶-N-氧化物与4-吡啶硼酸的摩尔比为1:2.2~2.5。
进一步优选的,步骤(2)中3,5-二溴吡啶-N-氧化物与4-吡啶硼酸的摩尔比为1:2.4。
优选的,所述Suzuki偶联反应中采用的碱为K2CO3。
进一步优选的,步骤(2)中3,5-二溴吡啶-N-氧化物与K2CO3的摩尔比为1:12.5。
一种金属有机框架Zn-MOF,其结构式为(ZnLBr2)n,其中,L为上述氧化吡啶配体L,n为非零的自然数。
优选的,所述金属有机框架是单斜晶系,属于P2(1)/n空间群。
进一步优选的,所述金属有机框架存在一种Zn(II)中心,其处于(ZnN2Br2)的配位环境中,和Zn(II)配位的氮原子是来自于配体L的端基吡啶,Zn-N键的长度分别是和其中,配体L中氧化吡啶的氧未参与配位,Zn(II)中心通过配体末端的吡啶氮原子相连,沿b轴方向伸展为一维螺旋链;一条链上氧化吡啶上的氧原子和另外一条链上氧化吡啶上的氢原子形成氢键,这些氢键将上述一维螺旋链进一步连接为波浪状的手性二维面。
更进一步优选的,两个Zn(II)之间的最短的距离是
一种金属有机框架Zn-MOF的制备方法,将ZnBr2溶液铺在上述氧化吡啶配体L溶液上,静置后得到无色单晶即为金属有机框架Zn-MOF。
优选的,所述ZnBr2溶液中的溶剂为甲醇。
优选的,所述ZnBr2溶液中ZnBr2的浓度为0.005mol/L。
优选的,所述氧化吡啶配体L溶液的溶剂为二氯甲烷。
优选的,所述氧化吡啶配体L溶液中氧化吡啶配体L的浓度为0.005mol/L。
优选的,ZnBr2与氧化吡啶配体L的投入摩尔比为1:1。
优选的,制备温度为室温。
本发明中所述的室温为15-25℃。
一种上述金属有机框架Zn-MOF在催化Knoevenagel缩合反应中的应用。
一种催化剂,包括上述金属有机框架Zn-MOF。
一种苄烯丙二腈的合成方法,以苯甲醛和丙二腈为原料,以上述金属有机框架Zn-MOF为催化剂,在室温下反应后即得苄烯丙二腈。
优选的,所述苯甲醛和丙二腈的摩尔比为1:1.2。
优选的,其步骤为,将苯甲醛与丙二腈混合搅拌一段时间后加入催化剂,继续搅拌一段时间后,即可获得苄烯丙二腈。
优选的,所述苯甲醛与催化剂的摩尔比为1:0.04。
一种上述金属有机框架Zn-MOF在催化Knoevenagel缩合反应后的回收方法,Knoevenagel缩合反应后,对混合液进行离心分离即可对金属有机框架Zn-MOF进行回收。
本发明的有益效果:
(1)采用本发明的Zn-MOF进行催化,实现了异相催化,同时制备的Zn-MOF稳定性好,可以重复利用五次以上,并且催化剂回收容易,提高了催化剂的利用率,降低了成本。
(2)本发明的反应温度温和,反应时间较短,活性高,催化剂用量少,无其他添加剂。
附图说明
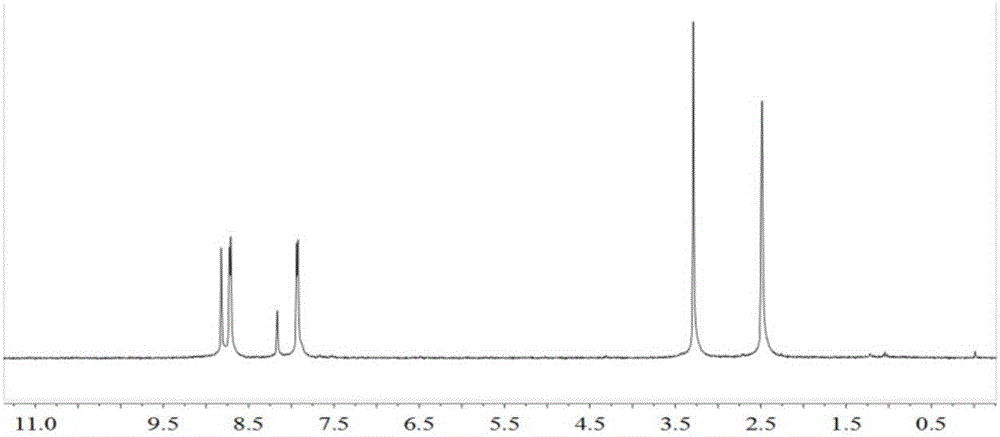
图1为本发明实施例1氧化吡啶配体L的1HNMR;
图2为本发明的氧化吡啶配体L的红外谱图;
图3为本发明的Zn-MOF的红外谱图;
图4为本发明的Zn-MOF的TGA谱图;
图5为本发明的Zn-MOF的单晶结构图、一维链状结构和手性二维面结构,其中,A为本发明的单晶结构图,B为本发明一维链状结构图,C为本发明手性二维面结构图;
图6为本发明的Zn-MOF在催化Knoevenagel缩合反应后的对应PXRD谱图。
具体实施方式
下面结合附图及实施例对本发明作进一步说明。
实施例1:氧化吡啶配体L的制备
具体的制备步骤如下:
(1)在250mL单口瓶中,将3,5-二溴吡啶(3.2g,13.5mmol)溶于50mL THF,加入间氯过氧苯甲酸(6.88g,40mmol),室温搅拌48小时,反应结束后加入饱和碳酸钠水溶液30mL,静置分层,水相用2×50mL CH2Cl2萃取,合并有机相并用无水Na2SO4干燥,硅胶柱层析(CH2Cl2)得浅白色固体1.53g,即为3,5-二溴吡啶-N-氧化物,产率44.1%。1H NMR(300MHz,DMSO,25℃,TMS):δ=8.60(s,2H,-C5H3NBr2),7.97(s,1H,-C5H3NBr2)。
(2)N2保护下,3,5-二溴吡啶-N-氧化物(2.53g,10.0mmol),4-吡啶硼酸(2.95g,24.0mmol),四三苯基磷合钯(1.16g,1.0mmol),无水碳酸钾(17.25g,125.0mmol),乙醇60mL,水60mL于三口瓶中,控温75℃,搅拌48h。减压蒸除溶剂,水洗,真空干燥,柱层析(CH2Cl2:CH3OH=20:1),得到白色粉末,即为氧化吡啶配体L。
对获得的氧化吡啶配体L进行核磁共振氢谱及红外谱图的表征,其结果如分别如图1、2所示。
实施例2:Zn-MOF的合成
在室温下,将ZnBr2(2.2mg,0.01mmol)溶于甲醇(2mL)获得ZnBr2的甲醇溶液,将L(2.4mg,0.01mmol)溶于CH2Cl2溶液(2mL)中,将甲醇溶液慢慢铺在CH2Cl2溶液上,静置三天后得到无色单晶。
通过红外谱图(IR),热重分析谱图(TGA)表征了该化合物,结果分别见图3、图4。
Zn-MOF单晶结构如图5所示。
由图5可以看出,Zn-MOF是单斜晶系,属于P2(1)/n空间群。Zn-MOF中只存在一种Zn(II)中心,其处于(ZnN2Br2)的配位环境中,和它配位的氮原子是来自于配体L的端基吡啶,Zn-N键的长度分别是和其中,配体L中氧化吡啶的氧未参与配位,Zn(II)中心通过配体末端的吡啶氮原子相连,沿b轴方向伸展为一维螺旋链。一条链上氧化吡啶上的氧原子和另外一条链上氧化吡啶上的氢原子形成氢键,这些氢键将上述一维螺旋链进一步连接为波浪状的手性二维面。Zn(II)和Zn(II)之间的最短的距离是具体晶体数据见表1
表1 Zn-MOF的晶体学数据
实验例3:苯甲醛与丙二腈缩合反应生成
室温下,在2mL玻璃瓶中加入苯甲醛(1mmol)与丙二腈(1.2mmol),搅拌5分钟后,加入催化剂Zn-MOF(19mg,4%(mol),以苯甲醛的量为基准),继续搅拌6h,用气相色谱追踪反应,反应结束后,快速离心,离心后的固体即为用于催化反应的催化剂Zn-MOF,回收催化剂,直接投入下一循环反应,按照上述条件,催化剂使用5个循环,反应液通过气相色谱计算产率,催化效果如表2所示,回收的催化剂通过PXRD表征,如图6所示。
表2 Zn-MOF催化Knoevenagel缩合反应5个循环的产率和TOF值
a:产率通过GC测定b:TOF=%yield(mmol of substrate/mmol of cat.h)
上述虽然结合附图对本发明的具体实施方式进行了描述,但并非对发明保护范围的限制,所属领域技术人员应该明白,在本发明的技术方案的基础上,本领域技术人员不需要付出创造性劳动即可做出的各种修改或变形仍在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!