有关病毒的无菌纯化方法与流程
本公开涉及用于纯化病毒的方法。
发明背景
管理机构,如世界卫生组织(worldhealthorganization)建立了有关制造如疫苗等施用给人类的药物组合物的标准和指导原则,其中限制了这些组合物的量和成分。例如,对于疫苗来说,有疫苗制剂必须是无菌的(即,不含独立复制的生物体)并且含有每次人用剂量不超过10ngdna等要求。为了确保供人类施用的组合物的安全性,这些标准应当遵守,而且可能在用于制造这些组合物的方法中带来挑战。
发明概要
本发明各方面提供了用于纯化病毒颗粒的方法,这些方法包括以下步骤:(a)提供含病毒颗粒的液体培养基,其中所述病毒颗粒的直径大于约100nm;(b)使所述病毒颗粒与固相基质接触,该固相基质包括配体活化的核及包含孔无活性壳,其中所述孔的分子量截留值阻止所述病毒颗粒进入所述配体活化的核中,并且其中小于所述孔的分子量截留值的分子能够进入所述配体活化的核中;及(c)通过过滤将所述固相基质与所述病毒颗粒分离以得到最终病毒制剂;其中所述方法是在无菌下执行。
在一些实施方案中,含病毒颗粒的液体培养基在步骤(b)之前经历一个或多个预纯化步骤。在一些实施方案中,预纯化步骤包括(a)通过酶处理来消化含多个病毒或病毒颗粒的液体培养基中的宿主细胞基因组dna;和/或(b)使用孔径等于或大于750kda的中空纤维膜对含多个病毒或病毒颗粒的液体培养基进行超滤/透滤。
在一些实施方案中,病毒颗粒的直径是约200nm、300nm、400nm、500nm或更大。在一些实施方案中,病毒是活病毒、减毒活病毒、修饰的活病毒或重组活病毒。在一些实施方案中,病毒属于选自由以下组成的组的病毒科:副粘病毒科(paramyxoviridae)、正粘病毒科(orthomyxoviridae)、黄病毒科(flaviviridae)、丝状病毒科(filoviridae)、沙粒病毒科(arenaviridae)、弹状病毒科(rhabdoviridae)及冠状病毒科(coronaviridae)。在一些实施方案中,病毒属于副粘病毒科(活病毒或灭活病毒)。在一些实施方案中,病毒是麻疹病毒。
在一些实施方案中,进入固相基质的核中的分子具有小于700kda的分子量。在一些实施方案中,固相基质中配体活化的核的配体能够通过阳离子相互作用、阴离子相互作用、疏水相互作用或混合相互作用结合进入该配体活化的核中的分子。在一些实施方案中,固相基质中配体活化的核的配体是辛胺。在一些实施方案中,固相基质是以浆液形式使用并且最终浓度介于2.5%(v/v)与30%(v/v)之间,优选是3.3%、5%、6.6%或10%,最优选是10%。在一些实施方案中,在搅拌下,在室温(20℃至25℃)下将固相基质与含病毒颗粒的液体培养基一起温育至少1小时,优选2小时、3小时或4小时,最优选2小时。
在一些实施方案中,相对于含多个病毒或病毒颗粒的液体培养基,最终病毒制剂的杂质相对减少60至95%。在一些实施方案中,最终病毒制剂的残留杂质小于1%。
在一些实施方案中,权利要求1中步骤(c)的过滤是使用孔径等于或大于1μm的过滤器进行。在一些实施方案中,在所述方法之后是一个或多个无菌过滤步骤。
在一些实施方案中,病毒是在选自由以下组成组的细胞系中繁殖:eb66细胞系、vero细胞系、vero-αhis细胞系、hela细胞系、hela-s3细胞系、293细胞系、pc12细胞系、cho细胞系、3t3细胞系、perc6细胞系、mdsk细胞系、鸡胚成纤维细胞系、鸭细胞系及二倍体禽类细胞系。在一些实施方案中,所述细胞系是鸭细胞系。在一些实施方案中,所述细胞系是二倍体禽类细胞系。在一些实施方案中,所述细胞系是eb66细胞系。
本发明各方面提供了任何本文所描述的方法的用途,这些方法用于制造供针对病毒感染免疫的组合物。
其它方面提供了包含通过本文所描述的任何方法可获得的病毒颗粒的组合物,这些组合物用于治疗和/或预防病毒感染。
附图简述
预期附图未按比例绘制。各图只是说明性的,而且并非实现本公开所需的。为清楚起见,在每个图中可能未标出每一成分。在附图中:
图1显示了麻疹病毒纯化方法的概述。
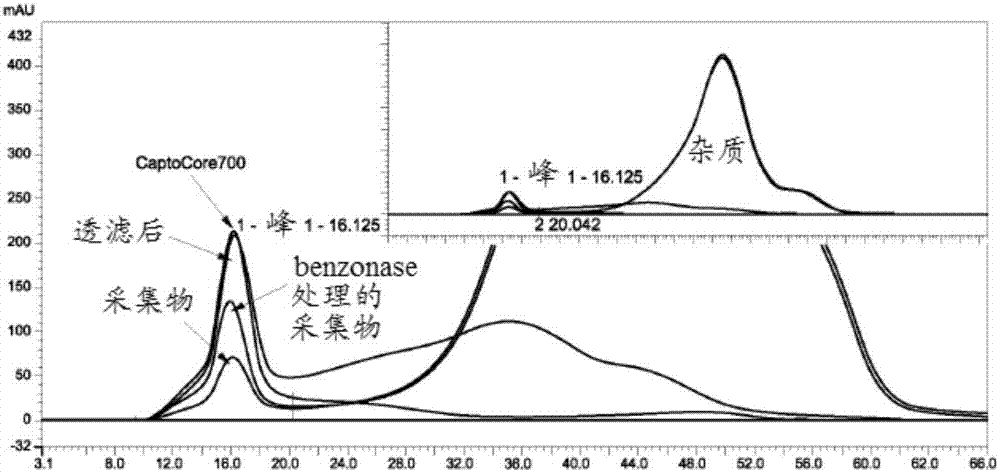
图2显示了在不同纯化步骤时麻疹病毒的尺寸排阻色谱法(sec)迹线。插图显示麻疹病毒(左侧峰)和杂质(右侧峰)的sec迹线。
图3a-3b显示在纯化过程各阶段期间样品中宿主细胞蛋白质(hcp)的存在情况。图3a显示了代表性银染色的sds-page凝胶。图3b显示了使用抗eb66-hcp-igg一次抗体的代表性western印迹。
图4a-4c显示了在透滤和
图5a和5b呈现了在纯化过程期间麻疹病毒的纳米颗粒跟踪分析(nta)。图5a显示来自麻疹病毒采集物的样品的nta。图5b显示经过透滤的样品的nta。
图6显示了来自麻疹病毒采集物(mv-gfp)的样品的尺寸排阻色谱图(荧光信号和uv214nm;右图)及由多角度静态光散射(mals)测定的病毒尺寸(左图)。
图7显示了高度纯化的麻疹病毒(mv-gfp)样品的尺寸排阻色谱图(荧光信号和uv214nm)及由多角度静态光散射(mals)测定的病毒尺寸(插图)。
图8呈现了在超滤/透滤以减少病毒制剂中的杂质之后间歇吸附的流程图。
图9显示了预先灭菌的加工组件的示意图。
图10呈现了在使用
图11呈现了在使用
图12显示了由尺寸排阻色谱法评估的间歇吸收样品中病毒(mv主峰回收率;菱形)和残留杂质(残余物回收率;正方形)的回收率百分比。
图13显示了经间歇吸附加工的样品中的残留杂质相对于未经过间歇吸附的样品相对减少。
图14呈现了经
图15显示了由尺寸排阻色谱法评估的在纯化过程期间的各个阶段经
图16显示了由sds-page凝胶评估的在纯化过程期间的各个阶段经
图17显示了由尺寸排阻色谱法评估的在纯化过程期间的各个阶段经
图18呈现的示意图显示了各种病毒科的分类和特性(http://www.nlv.ch/virologytutorials/classification.htm)。
发明详述
本文公开了用于纯化病毒的无菌方法及包含纯化的病毒的组合物。在制造施用给受试者的药物组合物,如疫苗组合物时,必须确保该组合物的无菌性和安全性。无菌组合物的制备典型地是在施用之前,通过对最终组合物进行无菌过滤,即,滤过0.2μm滤膜来实现。对于包含相对较大病毒(即,约100nm或更大)的组合物,如病毒疫苗组合物来说,无菌过滤可能由于病毒被截留在膜中而引起病毒的大量损失,由此降低纯化过程的病毒产率。另外,对剪切应力敏感的一些病毒,如麻疹病毒和其它病毒来说,纯化过程的每个步骤必须使用轻柔条件进行,由此使替代性灭菌方法无用。已发现,使用切向流过滤的病毒纯化方法并不成功,也不能足够有效地制备如麻疹病毒等相对较大病毒的无菌组合物(参见美国专利号7,759,104及其中的实施例)。此外,保持和实现切向流过滤系统无菌很昂贵,而且需要大量的付出。本文所描述的方法允许在完全无菌条件下进行病毒制备,由此使病毒制备不需要无菌过滤。
本发明的应用不局限于以下描述中陈述或附图中说明的构造细节和组分布置。本发明能够包含其它实施方案,并且能够以各种方式实践或进行。另外,本文中使用的措辞和术语都是出于描述的目的,而不应视作限制。本文中使用的“包括”、“包含”或“具有”、“含有”、“涉及”等旨在涵盖其后所列各项和其等效物以及其它各项。
本发明各方面涉及用于纯化病毒的方法。希望用于纯化病毒制剂的任何病毒都可以与本发明各方面相容。术语“病毒”、“病毒颗粒”、“病毒的颗粒”及“病毒体”可以互换使用并且意思指包含由蛋白质衣壳(壳体)及任选地脂质包膜包围的遗传物质的病毒。一般来说,病毒可以基于蛋白质衣壳内所包含的病毒遗传物质及病毒能够在感染细胞(宿主细胞)中产生信使rna(mrna)所凭借的方法进行分类,参见图18。举例来说,病毒可以是dna病毒或rna病毒。在一些实施方案中,病毒是逆转录病毒,意思指病毒在复制期间通过中间物逆转录其核酸。在一些实施方案中,病毒是双链dna(dsdna)病毒、单链dna(ssdna)病毒、双链rna(dsrna)病毒、正链单链rna(+ssrna)病毒、负链单链rna(-ssrna)病毒、单链rna逆转录病毒(ssrna-rt)或双链dna逆转录病毒(dsdna-rt)。
病毒还可以基于它能够感染的宿主细胞的类型进行分类。如本文所使用,如果病毒能够进入细胞,复制并且从细胞释放,则该病毒能够感染该细胞。在一些实施方案中,病毒可以是死病毒或灭活病毒。在此类实施例中,如果病毒不是死病毒或灭活病毒,则认为该病毒能够感染该病毒能够进入、复制并且被释放的宿主细胞。在一些实施方案中,病毒能够感染真核细胞。在一些实施方案中,病毒是动物病毒(即,能够感染动物细胞)。在其它实施方案中,病毒是植物病毒(即,能够感染植物细胞)。
本文所描述的方法可以用于纯化活病毒或者死病毒或灭活病毒。在一些实施方案中,病毒是减毒活病毒。举例来说,相较于野生型病毒,该病毒可以在宿主中具有减低的感染性、毒力和/或复制。在其它实施方案中,相较于野生型病毒,该病毒可以在宿主中具有增强的感染性、毒力和/或复制。在一些实施方案中,病毒是突变或修饰的病毒,例如相对于野生型病毒,该病毒的核酸可以含有至少一个突变。在一些实施方案中,病毒是重组活病毒,意思指病毒是重组产生的并且可以含有来自不同来源的核酸。在一些实施方案中,病毒对剪切应力敏感。在一些实施方案中,轻柔地处理病毒以减少病毒滴度和感染性损失。
一般来说,病毒的尺寸范围是约20nm至超过1μm长度。如本文所描述,涉及经由0.2μm过滤(“无菌过滤”)的病毒制备方法,特别是施用给受试者的病毒的制备方法,很难或者不能用于约100nm或比过滤器孔径大的病毒。本文所描述的方法可以用于纯化任何病毒,但可能特别适用于尺寸是约100nm或更大的病毒。在一些实施方案中,病毒的平均尺寸是约100nm、150nm、200nm、250nm、300nm、350nm、400nm、450nm、500nm、550nm、600nm、650nm、700nm、750nm、800nm、850nm、900nm、950nm或约1000nm或者更大。在一些实施方案中,病毒可以是多形性病毒,意味着在病毒群内,各病毒可以呈不同尺寸和/或形状。在一些实施方案中,病毒属于副粘病毒科、正粘病毒科、黄病毒科、丝状病毒科、沙粒病毒科、弹状病毒科或冠状病毒科。在一些实施方案中,病毒是来自逆转录病毒科、冠状病毒科、丝状病毒科、弹状病毒科、布尼亚病毒科(buyna)、正粘病毒科、副粘病毒科、沙粒病毒科、疱疹病毒科、虹彩病毒科、杆状病毒科或痘病毒科的病毒。用于本文所描述的方法的特别优选的病毒是麻疹病毒、hiv、巨型模仿病毒(giantmimivirus)或疱疹病毒。在一些实施方案中,病毒是减毒形式,如任何本文所描述的病毒的减毒病毒,例如减毒的麻疹病毒。
本文所描述的本发明各方面涉及用于纯化病毒的无菌方法。如本文所使用,术语“无菌”是指组合物、方法及条件不含任何污染性活生物体。在一些实施方案中,该方法的各个步骤都是在无菌条件下执行,由此使所得到的病毒制剂可以不含独立复制的活生物体。
本文所描述的方法提供了通过连续步骤去除病毒制剂中的杂质或污染物来纯化病毒的方法。如本文所使用,“杂质”和“污染物”可以互换使用并且是指在纯化过程期间的任何步骤时病毒制剂中的不想要成分。在一些实施方案中,杂质或污染物可以是宿主细胞或其片段,包括宿主细胞dna和/或宿主细胞蛋白质;病毒片段或病毒核酸;酶,如
本文所描述的方法涉及提供含多个供纯化的病毒的液体培养基。病毒可以由本领域中已知的任何方法制造或提供。举例来说,可以通过在活宿主、胚胎、组织培养物,或细胞系,如
在某些实施方案中,使病毒在细胞或组织培养物中繁殖。允许病毒进入和复制(能够被病毒感染)的任何细胞都可以用于病毒繁殖。在一些实施方案中,细胞是原代细胞(例如从宿主生物体分离的细胞)。在一些实施方案中,细胞是来自细胞系。在一些实施方案中,细胞系是来源于哺乳动物(如人类或非人类哺乳动物)、鸟类、昆虫或植物的细胞。在一些实施方案中,细胞系的细胞是
病毒在细胞或细胞群中复制之后,病毒可以被释放至受感染细胞周围的液体培养基中。在一些实施方案中,宿主细胞可以被溶解(例如以酶方式、机械方式)以将病毒释放至液体培养基中。释放入病毒的液体培养基的类型将取决于宿主细胞的类型及所用病毒繁殖方法。在一些实施方案中,液体培养基含有血清、血浆、血液、细胞外液、羊水、卵黄囊、缓冲液,或者细胞或组织培养基。支持细胞或细胞群生长的任何细胞或组织培养基都可以使用。
在一些实施方案中,细胞在培养基质,如烧瓶、培养皿或培养盘上以单层形式生长。在此类实施方案中,通过从细胞去除培养基,从细胞采集病毒。在一些实施方案中,将细胞溶解以将病毒释放至培养基中并收集培养基以采集病毒。在其它实施方案中,细胞是悬浮生长的,在此情况下细胞是漂浮的或仅轻轻地粘附于培养基质。在一些实施方案中,培养基质可以是滚瓶、摇瓶、旋转瓶或生物反应器。在又其它实施方案中,细胞以混合培养方式生长,在此情况下,一部分细胞粘附于培养基质,而一部分细胞是漂浮并且不粘附的。在一些实施方案中,细胞和病毒都存在于液体培养基中。
细胞培养方法是本领域技术人员显而易见的。参见例如,generaltechniquesofcellculture,cambridgeuniversitypress,cambridge,unitedkingdom。
在一些实施方案中,含病毒的液体培养基经历一个或多个预纯化步骤。在一些实施方案中,可以使用一个或多个预纯化步骤以例如减少一种或多种杂质或污染物的存在,去除宿主细胞或其片段,增加病毒产率,和/或缩短总加工时间。
在一些实施方案中,可以通过本领域中已知的任何适合方式将任何宿主细胞或其片段从含病毒的液体培养基分离或去除。在一些实施方案中,宿主细胞是通过对液体培养基进行离心或过滤来去除。离心可以在能够将宿主细胞或其片段与病毒分离的速度和持续时间下进行。举例来说,宿主细胞或其片段形成团粒,而病毒保留在液体培养基中。替代地或另外,可以使用过滤方法,如膜过滤,将宿主细胞或其片段从含病毒的液体培养基去除(例如超滤)。在一些实施方案中,滤膜的选择使得病毒能够穿过过滤器,而宿主细胞或其片段被截留在膜中。
在一些实施方案中,所述一个或多个预纯化步骤涉及降解含病毒的液体培养基中的宿主细胞基因组dna。在一些实施方案中,通过酶处理使宿主细胞基因组dna降解。任何dna降解酶都可以与本文所描述的方法相容。在一些实施例,该酶是核酸酶。在一些实施方案中,该核酸酶降解dna和rna两种。核酸酶的非限制性实例包括但不限于,
在一些实施方案中,所述一个或多个预纯化步骤涉及对含病毒的液体培养基进行超滤和/或透滤。如本文所使用,“超滤”是指基于各成分的尺寸或分子量,通过使液体培养基穿过半透膜来分离混合物中各成分的一种方法。分子量大于半透膜孔径(分子量截留值(mwco))的成分被保留在膜上,而较小分子量的成分被允许穿过该膜。如本文所使用,“透滤”是指降低混合物中的成分,如杂质或污染物的浓度,和/或交换缓冲液的一种方法。透滤可以通过多种方法进行,例如连续透滤、不连续透滤或依序透滤。在一些实施方案中,同时或依序进行超滤法和透滤法。
在一些实施方案中,超滤和透滤是使用切向流过滤进行的。如本文所使用,“切向流过滤”,又称为“错流过滤”,是进料流(即,含病毒的液体培养基)与滤膜相切的一种过滤方法。在一些实施方案中,切向流过滤是使用中空纤维膜进行的。将进料流馈送至管状纤维中并且进料中小于膜mwco的成分被允许穿过并离开该物流,而较大成分被保持在该物流中并且可以通过系统再循环。额外液体培养基或替代缓冲液可以在与混合物中较小成分离开相同的速率下连续地添加至该物流中,由此维持恒定的病毒浓度。在一些实施方案中,含病毒的液体培养基经历至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29体积,或至少30体积的液体培养基或替代缓冲液交换。替代缓冲液的非限制性实例包括磷酸盐缓冲溶液(pbs)、dulbecco磷酸盐缓冲生理盐水(dpbs)、earle平衡盐溶液(ebss)、hank平衡盐溶液(hbss),或水。
在一些实施方案中,膜的mwco是至少500kda(千道尔顿)、510kda、520kda、530kda、540kda、550kda、560kda、570kda、580kda、590kda、600kda、610kda、620kda、630kda、640kda、650kda、660kda、670kda、680kda、690kda、700kda、710kda、720kda、730kda、740kda、750kda、760kda、770kda、780kda、790kda、800kda、810kda、820kda、830kda、840kda、850kda、860kda、870kda、880kda、890kda或至少900kda。在一些实施方案中,膜的mwco大于或等于750kda。
本公开各方面涉及使含病毒的液体培养基与固相基质接触。在一些实施方案中,通过间歇吸附使含病毒的液体培养基与固相基质接触。如本文所使用,“间歇吸附”是指将固相基质添加至包括希望纯化的分子(例如病毒)的成分的液相混合物(例如含病毒的液体培养基)中的一种方法。在一些实施方案中,将固相基质悬浮于缓冲溶液中,称为浆液。固相基质吸附该混合物的成分。随后,可以使用本领域中已知的任何方法,如离心、过滤或絮凝,将固相基质和吸附的成分与该混合物分离。在一些实施方案中,希望纯化的分子(例如病毒)被吸附至固相基质上。在其它实施方案中,杂质或污染物被吸附至固相基质上并且希望纯化的分子保留在液相中。通用间歇吸附方法和考虑因素可以见于例如proteinpurification:principlesandpractice,第3版,springeradvancedtextsinchemistry,newyork,ny中。
在一些实施方案中,固相基质包含基质及结合混合物各成分的配体。在一些实施方案中,该基质是
在一些实施方案中,固相基质的配体是辛胺、二乙氨基乙基、季铵或磺酸酯。可以与本文所描述的方法相容的固相基质的非限制性实例包括但不限于,
在一些实施方案中,固相基质在与含病毒的液体培养基组合之前,悬浮于缓冲溶液中呈浆液形式。在一些实施方案中,将呈浆液形式的固相基质以介于2.5%(v/v)-30%(v/v)之间、介于5%(v/v)-20%(v/v)之间或介于7.5%(v/v)-15%(v/v)之间的最终浓度与含病毒的液体培养基组合。在一些实施方案中,以约2.5%、3.0%、3.5%、4.0%、4.5%、5.0%、5.5%、6.0%、6.5%、7.0%、7.5%、8.0%、8.5%、9.0%、9.5%、10%、10.5%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、21%、22%、23%、24%、25%、26%、27%、28%、28%、29%或30%(v/v)的最终浓度添加该浆液。在一些实施方案中,以约10%(v/v)的最终浓度添加该浆液。
为了增加病毒的回收率并且增进液体培养基中杂质的结合和去除,可以改变条件,包括固相基质与含病毒的液体培养基之间的接触的持续时间、温度及模式。在一些实施方案中,固相基质与含病毒的液体培养基是在介于15℃-30℃之间,如17℃-27℃或20℃-25℃的温度下接触或温育。在一些实施方案中,固相基质与含病毒的液体培养基是在室温下接触或温育。在一些实施方案中,固相基质与含病毒的液体培养基是在15℃、16℃、17℃、18℃、19℃、20℃、21℃、22℃、23℃、24℃、25℃、26℃、27℃、28℃、29℃或30℃温度下接触或温育。
在一些实施方案中,固相基质与含病毒的液体培养基接触或一起温育的持续时间在1小时与5小时之间、在1小时与10小时之间、在1小时与24小时之间、在5小时与10小时之间、在10小时与15小时之间,或在15-24小时之间。在一些实施方案中,固相基质与含病毒的液体培养基接触或一起温育约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23或24小时。在一些实施方案中,固相基质与含病毒的液体培养基接触或一起温育约2小时。
在任何本文所描述的实施方案中,可以通过本领域中已知的任何模式使固相基质与含病毒的液体培养基接触或一起温育。举例来说,固相基质与含病毒的液体培养基可以在容器中静态地或在振荡、倒转、摇动或搅拌下接触或温育。在一些实施方案中,固相基质与含病毒的液体培养基是在搅拌下温育。
在间歇吸附之后,通过本领域中已知的任何方法,如离心、过滤或絮凝,将固相基质和任何结合的成分从液相去除。在一些实施方案中,通过过滤,如通过本文所描述的任何过滤方法去除固相基质和任何结合的成分。在一些实施方案中,使用孔径是至少0.5μm、0.6μm、0.7μm、0.8μm、0.9μm、1μm、1.1μm、1.2μm、1.3μm、1.4μm、1.5μm、1.6μm、1.7μm、1.8μm、1.9μm或至少2.0μm的膜,通过膜过滤去除固相基质和任何结合的成分。在一些实施方案中,膜的孔径大于或等于1.0μm。本文所描述的方法中使用的固相基质可以再生(例如清洁和再灭菌)并再次用于间歇吸附。
使用任何本文所描述的方法制造的病毒制剂可以进一步经历额外加工步骤,包括额外的过滤步骤和/或冻干。病毒制剂还可以经历制剂纯度分析。举例来说,还可以评估病毒制剂中杂质和污染物、宿主细胞基因组dna和/或宿主细胞蛋白质的存在。可以使用本领域中已知的任何方法,如尺寸排阻色谱法(sec)、在不同波长下的光学密度、蛋白质凝胶电泳(例如sds-page)、western印迹、elisa、pcr和/或qpcr评估病毒制剂的纯度。
在一些实施方案中,评估病毒制剂中残留杂质或污染物的量。在一些实施方案中,将残留杂质或污染物的量与在纯化方法早期杂质或污染物的量相比较。在一些实施方案中,相对于在纯化方法早期杂质的存在,最终病毒制剂中杂质相对减少60-95%。在一些实施方案中,最终病毒制剂中杂质相对减少约60%、61%、62%、63%、64%、65%、66%、67%、68%、69%、70%、71%、72%、73%、74%、75%、76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%或95%。在一些实施方案中,最终病毒制剂含有低于5%的杂质或污染物。在一些实施方案中,最终病毒制剂含有低于5%、4%、3%、2%、1%、0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%或低于0.1%的杂质。在一些实施方案中,最终病毒制剂含有低于1%的杂质。
任何本文所描述的方法都可以用于制造供施用给受试者的含纯化病毒的组合物。在一些实施方案中,受试者是哺乳动物受试者,如人类或非人类动物,包括家畜、宠物或伴侣动物。在一些实施方案中,该组合物可以施用给需要针对病毒制剂中的病毒或类似病毒进行免疫的受试者。在一些实施方案中,包含使用本文所描述的方法纯化的病毒的病毒制剂或组合物用于治疗或预防病毒制剂中的病毒或类似病毒的感染。
使用本文所描述的方法纯化的病毒制剂或组合物病毒可以通过本领域中已知的任何途径施用给受试者。在一些实施方案中,这些制剂或组合物可以通过常规途径,如经肠胃外施用。如本文所使用,“肠胃外”施用包括但不限于,皮下、皮内、静脉内、肌肉内、动脉内、鞘内或通过输注施用。
除非本文另作定义,否则结合本公开使用的科技术语应当具有与本领域普通技术人员通常所了解的含义。另外,除非上下文另外需要,否则单数的术语应包括复数形式,而且复数的术语应包括单数形式。本公开的方法和技术一般是根据本领域中众所周知的常规方法进行。一般来说,结合本文所描述的生物化学、酶学、分子和细胞生物学、微生物学、病毒学、细胞或组织培养、遗传学以及蛋白质和核酸化学使用的命名法,以及生物化学、酶学、分子和细胞生物学、微生物学、病毒学、细胞或组织培养、遗传学以及蛋白质和核酸化学的技术是本领域中众所周知并且常用的那些。除非另作指示,否则本公开的方法和技术一般是根据本领域中众所周知的常规方法并且如本说明书全篇所引用和论述的各种通用和更具体的参考文献中所描述来进行。
本发明将借助于以下实施例进一步说明,这些实施例不应被解释为进一步限制本发明。本申请通篇引用的所有参考资料(包括参考文献、颁布的专利、公布的专利申请及共同待决的专利申请)的全部内容都以引用的方式清楚地并入本文中,特别是有关以上所提到的传授内容。不过,任何参考文献的引用都不打算承认该参考文献是现有技术。
实施例
实施例1:有关在eb66细胞中制造的活麻疹病毒疫苗的纯化方法的开发
开发针对在
受感染细胞的培养基的澄清
在细胞培养中产生病毒之后,通过过滤去除细胞。针对在病毒颗粒无明显损失下细胞培养物上清液中细胞的去除情况来评价深度过滤和膜过滤以及适合的分离范围(深度过滤)和孔径(膜过滤)。使用含3μm/1μm(parkerplplk-01dd-pnl-s)的组合过滤器分离细胞与病毒颗粒。过滤后,通过目测显微镜分析在澄清上清液中未检测到
dna去除
使用内切核酸酶
在室温(18-22℃)过夜温育(约12-16小时)时使用5u/ml的最终酶浓度进行dna降解。利用qpcr针对90bp、176bp及316bp扩增子分析残留dna含量和尺寸。使用这些方法,将宿主细胞dna(hcd)中尺寸大于317bp的dna的总量从440ng降低至0.5ng并且尺寸大于176bp的dna的总量从1400ng降低至2ng(表1)。
通过切向流过滤纯化麻疹病毒
应用基于中空纤维的切向流过滤(tff)系统及相应过程参数使剪切力减到最小并且避免病毒包膜的破坏和病毒感染性的损失。发现各种管腔直径(0.5至1.0mm)及再循环流动速率都具有在1000至6000s-1之间的剪切速率。对膜截留值进行优化以允许完全截留麻疹病毒,同时使各种杂质(蛋白质、dna片段、培养基成分)穿过该膜。该过程是在室温下进行,因此过程时间也应予以考虑。
使用750kda中空纤维膜(gehealthcare),在对应于2000s-1剪切速率的流动速率下,将澄清并且经过
通过间歇吸附纯化麻疹病毒
开发可以在超滤/透滤步骤后应用的可选间歇吸附加工步骤。评估利用不同配体(阳离子、阴离子、疏水及混合模式)的各种色谱树脂与残留杂质(例如hcp、dna片段)的结合,同时麻疹病毒被保留在上清液中。使用
通过过滤进行最终精制
最后一个精制步骤是在存在或不存在
表1:整个纯化过程中杂质的去除情况
方法
尺寸排阻色谱法
使用尺寸排阻色谱法(sec),相对于也产生uv吸收的杂质(例如hcp、dna)测定在整个纯化过程中病毒的纯度。图2和表2提供了在整个纯化过程中病毒纯度的近似值。观察到杂质(主要是宿主细胞蛋白质)明显减少。
应用sec加上uv吸收(在不同波长下)和荧光发射(gfp特有的)进行病毒分析。简单地说,在sephacryls500色谱柱(10×300mm;分离范围是至多20mioda)上,利用补充有250mm氯化钠的pbs缓冲液以0.5ml/min流动速率进行分离。记录在214nm、280nm及260nm下的uv信号。在em509nm(ex395nm)波长下检测gfp特有的荧光。通过将sec连接至多角度静态光散射(mals)检测器来收集有关病毒尺寸的额外数据。利用
表2:图2中呈现的迹线的尺寸排阻色谱分析
sds-page和western印迹
除定量elisa外,还可以利用sds-page和western印迹法定性地监测整个纯化过程中宿主细胞蛋白质的存在和减少情况,参见(图3a、3b、4a及4b)。另外,使用特异性抗体检测病毒蛋白质的存在(图4c)。
简单地说,在200v下,在4-12%bistris凝胶上分离减少的样品50分钟。将银染色或未染色的凝胶转印至硝基纤维素膜上进行western印迹。利用适当抗体(分别是抗
纳米颗粒跟踪分析
使用纳米颗粒跟踪分析(nta),利用nanosight仪器测定病毒颗粒的浓度和尺寸。在下游加工(dsp)期间颗粒尺寸的变化可以指示病毒颗粒完整性的损失。根据nta,发现病毒体的颗粒尺寸在约100-400nm范围内(图5a-5b)。尺寸分布不均匀,但显示出明显的多形现象(根据文献),而且在整个纯化中几乎保持恒定。如图5a-5b中所示,采集物及透滤后的跟踪分析图显示,颗粒尺寸分布无明显变化。
宿主细胞dna(hcd)的定量
利用qpcr测定来自
评估宿主细胞蛋白质
利用elisa确定来自
残留benzonase检测
使用可商购的elisa试剂盒(来自merck的benzonaseelisaii,产品号1.01681)测定残留
纯化方法mv-gfp显示有效去除宿主细胞dna。采集物的起始宿主细胞dna含量对于176bp扩增子是1400ng/ml并且对于317bp扩增子是440ng/ml。通过benzonase处理有效去除宿主细胞dna并且在后续纯化步骤中几乎保持恒定。根据针对176bp扩增子的qpcr分析,在超滤/透滤后的残留宿主细胞dna浓度小于4ng/ml。如通过317bp扩增子所分析,较大dna片段是约1ng/ml。应用分批色谱法未能进一步降低残留dna的含量。将施用给受试者的最终组合物调整到每剂至少103tcid50。可能需要进一步稀释病毒制剂(约106tcid50)并且将宿主细胞dna的最终含量降低至每剂约0.4至0.04ng的估计量。
利用elisa测得宿主细胞蛋白质的最终浓度小于10μg/ml,该浓度也进一步降低至每剂小于1μg。使用
实施例2:间隙吸附色谱法步骤的优化
为了在超滤/透滤步骤后进一步减少mv-gfp材料中的残留杂质,开发出利用色谱树脂作为吸附剂的间歇吸附法。间歇吸附是一个单阶段步骤,并且涉及在适合混合容器中将吸附剂(树脂)添加至mv-gfp材料中并温育指定时间段。随后,例如通过过滤或离心来去除吸附剂。可以在利用高压釜对吸附剂灭菌后,将吸附剂以无菌方式引入加工袋中。方法流程图显示于图8中。加工组件,包括间歇吸附,绘于图9中。
筛选用于间歇吸附的树脂
为了鉴别适合减少残留杂质的树脂,小规模筛选利用不同配体(阳离子、阴离子、疏水及混合模式)的各种色谱树脂。测试的树脂包括
这些不同的色谱树脂分别在pbs中制备成50%浆液,并且在超滤/透滤后以10%v/v添加至mv-gfp采集物中。在4℃下温育所有样品过夜。通过离心去除树脂。如实施例1中所描述,利用尺寸排阻色谱法分析上清液。
表3:筛选色谱树脂用于选择固相基质
另外,对树脂组合进行评价,包括
另外,对添加至病毒样品中的
下游加工的开发
在优化
相较于未用间歇吸附加工的材料,杂质相对减少66至93%。mv制剂的纯度在50%至76%范围内并且在用20个透滤循环(缓冲液体积交换)结合间歇吸附加工的样品中实现。用间歇吸附加工的材料的病毒产率与未用间歇吸附加工的相应材料的产率相当。
过滤采集材料并用50单位的
经历间歇吸附的样品以及未经历间歇吸附的样品中mv-gfp和残留杂质的回收率呈现于图12中。经历间歇吸附的样品相对于未经历间歇吸附的样品的残留杂质的相对减少显示于图13中。尽管相对mv-gfp回收率在所有样品中保持恒定,但使用
利用由尺寸排阻色谱法得到的数据计算从经历和不经历间歇吸附加工的样品回收的mv-gfp的总产率(图14)。20个透滤循环后的mv-gfp产率在未经历间歇吸附加工的样品中是51%,并且在经历间歇吸附加工的样品中是54%。
使用尺寸排阻色谱法定量评估并且使用sds-page电泳定性评估在每个加工阶段之后mv-gfp材料的纯度。mv-gfp材料的纯度在间歇吸附之后明显提高(图15和图16)。尽管利用20dv样品在无间歇吸附情况下达到29%的最大纯度,但在仅5个透滤体积交换(5dv)加上间歇吸附之后就达到相当的纯度。由此使加工时间明显缩短。利用20dv样品加上间歇吸附达到最高纯度水平(69%)。
图17显示,经历和未经历间歇吸附加工的样品中的残留杂质相较于浓缩的采集物样品的减少。在无间歇吸附情况下,残留杂质减少96%或log1.4。使用
总的说来,使用
表4:有关mv间歇吸附实验的概述
- 还没有人留言评论。精彩留言会获得点赞!