含有反向取向人泛素C启动子的逆转录病毒载体的制作方法
相关申请的交叉引用
本申请要求2015年7月1日提交的ussn62/187,678的优先权和权益,所述专利出于所有目的以引用的方式整体并入本文。
政府资助的声明
本发明根据国立卫生研究院的国家心肺及血液研究所(nationalheartlungandbloodinstituteatthenationalinstitutesofhealth)授予的批准号hl073104在政府支持下进行。政府享有本发明中的某些权利。
背景
基因递送至人细胞已被探索作为校正或保护免受各种人疾病如先天性酶缺陷或恶性血液病的遗传改变的手段。已经开发了各种基因转导系统,包括致癌逆转录病毒载体(oncoretroviralvector)、慢病毒载体、腺病毒载体和腺相关病毒载体。然而,尽管载体系统多种多样,但细胞转导效率对于治疗功效仍然可能太低。
尽管对有效载体有公认需要,但大多数靶细胞中转基因的高水平表达对基因转移技术而言一直是重大挑战。
概述
在各个实施方案中,提供了包含处于反向取向的人泛素c启动子的逆转录病毒载体,以及含有此类载体的病毒颗粒、用此类载体转导的宿主细胞和利用此类载体的治疗方法。
本文设想的各个实施方案可包括但不必限于以下中的一个或多个:
实施方案1:一种重组逆转录病毒载体,所述载体包含人泛素c(ubc)启动子和多克隆位点,其中所述ubc启动子在所述载体中处于反向取向,以使得自所述启动子的转录方向朝向所述载体的5'长末端重复序列(ltr)定向并转录插入所述多克隆位点中的核酸。
实施方案2:一种重组逆转录病毒载体,所述载体包含可操作地连接至转基因的人泛素c(ubc)启动子,其中所述启动子和所述转基因处于反向取向,以使得所述转基因自所述启动子转录的方向朝向所述载体的5'长末端重复序列(ltr)定向。
实施方案3:根据实施方案1至2中任一项所述的载体,其中所述启动子包括来自人泛素c基因ucsc人基因组序列型式hg19减去约125398318位至约125399530位的链的片段或由所述片段组成。
实施方案4:根据实施方案1至3中任一项的载体,其中所述启动子内的内含子在逆转录病毒包装期间不丢失。
实施方案5:根据实施方案1至4中任一项所述的载体,其中所述载体含有处于反向取向的聚腺苷酸化信号。
实施方案6:根据实施方案5所述的载体,其中所述聚腺苷酸化信号(polya)被插入所述启动子的3',所述3'是所述启动子的相对于整个载体序列的5'。
实施方案7:根据实施方案5至6中任一项所述的载体,其中所述聚腺苷酸化信号选自由以下各项组成的组:牛生长激素聚腺苷酸化信号序列、人生长激素聚腺苷酸化信号、兔β-球蛋白基因聚腺苷酸化信号、人疱疹病毒(hsv)聚腺苷酸化信号、胸苷激酶(tk)基因聚腺苷酸化信号、以及源自现有基因组或计算机设计并合成的其他信号。
实施方案8:根据实施方案5至6中任一项所述的载体,其中所述聚腺苷酸化信号是牛生长激素聚腺苷酸化信号序列或人生长激素聚腺苷酸化信号。
实施方案9:根据实施方案1至8中任一项所述的载体,其中与具有处于非反向取向的ubc启动子的相同载体相比,所述载体在瞬时转染和稳定转导的细胞系中提供表达的至少约2倍增加。
实施方案10:根据实施方案1至9中任一项所述的载体,其中与具有处于非反向取向的ubc启动子的相同载体相比,所述载体在转导的原代细胞中提供表达的至少约4倍增加。
实施方案11:根据实施方案1至10中任一项所述的载体,其中所述逆转录病毒载体选自由以下各项组成的组:hiv-1慢病毒载体、hiv-2慢病毒载体、α逆转录病毒载体、马传染性贫血病毒(eiav)慢病毒载体、momlv载体、x-mlv载体、p-mlv载体、a-mlv载体、galv载体、hev-w载体、siv-1载体、fiv-1载体以及serv-1-5载体。
实施方案12:根据实施方案11所述的载体,其中所述逆转录病毒载体是慢病毒载体。
实施方案13:根据实施方案12所述的载体,其中所述逆转录病毒载体是基于hiv-1的慢病毒载体。
实施方案14:根据实施方案12至13中任一项所述的载体,其中所述慢病毒载体是非tat依赖性和自灭活(sin)慢病毒载体。
实施方案15:根据实施方案1至14中任一项所述的载体,其中所述载体是双向载体。
实施方案16:根据实施方案1至15中任一项所述的载体,其还包含在所述3'ltr中的绝缘子。
实施方案17:根据实施方案16所述的载体,其中所述绝缘子包含77bp绝缘子元件fb(fii/bead-a),所述绝缘子含有鸡β-球蛋白5'dnasei超敏位点4(5'hs4)的最小ctcf结合位点增强子阻断组分。
实施方案18:根据实施方案1至17中任一项所述的载体,其中所述载体包含ψ区载体基因组包装信号。
实施方案19:根据实施方案1至18中任一项所述的载体,其中所述载体包含rev响应元件(rre)。
实施方案20:根据实施方案1至19中任一项所述的载体,其中所述载体包含中央聚嘌呤段(centralpolypurinetract)。
实施方案21:根据实施方案1至20中任一项所述的载体,其中所述载体包含翻译后调控元件。
实施方案22:根据实施方案21所述的载体,其中所述转录后调控元件是修饰的土拨鼠转录后调控元件(wpre)。
实施方案23:根据实施方案1至22中任一项所述的载体,其中所述载体不能通过重组来重构野生型慢病毒。
实施方案24:根据实施方案2至23中任一项所述的载体,其中所述载体包含可操作地连接至所述ubc启动子的转基因,其中所述转基因表达用于治疗选自由以下各项组成的组的病变的基因产物:scid、镰状细胞病、脂质体贮积症、囊性纤维化、肌营养不良、苯丙酮尿症、帕金森氏病和血友病。
实施方案25:根据实施方案2至15中任一项所述的载体,其中所述载体表达一种或多种选自由以下各项组成的组的基因产物:腺苷脱氨酶(ada)、il-2受体γ(il-2rγ)、嘌呤核苷磷酸化酶(pnp)基因、janus激酶-3(jak3)、artemis基因、抗镰状化人β-球蛋白基因、因子viii、因子ix、cftr、全长或缩短的肌营养不良蛋白、abcd1基因、th、aadc、和gch1、天冬氨酰基氨基葡糖苷酶、α-半乳糖苷酶a、棕榈酰基蛋白质硫酯酶、三肽基肽酶、溶酶体跨膜蛋白、半胱氨酸转运蛋白、酸性神经酰胺酶、酸性α-l-岩藻糖苷酶、保护性蛋白/组织蛋白酶a、酸性β-葡糖苷酶、酸性β-半乳糖苷酶、艾杜糖醛酸-2-硫酸酯酶、α-l-艾杜糖醛酸酶、半乳糖脑苷酯酶、酸性α-甘露糖苷酶、酸性β-甘露糖苷酶、芳基硫酸酯酶b、芳基硫酸酯酶a、n-乙酰基半乳糖胺-6-硫酸酯、酸性β-半乳糖苷酶、n-乙酰基葡糖胺-1-磷酸转移酶、酸性鞘磷脂酶(asm)、npc-1、α-葡糖苷酶、β-氨基己糖苷酶b、乙酰肝素n-硫酸酯酶、α-n-乙酰基氨基葡糖苷酶、乙酰辅酶a:α-氨基葡糖苷、n-乙酰基葡糖胺-6-硫酸酯、α-n-乙酰基氨基半乳糖苷酶、α-n-乙酰基氨基半乳糖苷酶、α-神经氨酸酶、β-葡糖苷酸酶、β-氨基己糖苷酶a、酸性脂肪酶。
实施方案26:根据实施方案24所述的载体,其中所述转基因表达用于治疗ada-scid的腺苷脱氨酶(ada)。
实施方案27:如实施方案24所述的载体,其中所述转基因表达用于治疗x-scid的il-2受体γ(il-2rγ)基因/cdna。
实施方案28:如实施方案24所述的载体,其中所述转基因表达抗镰状化人β-球蛋白基因。
实施方案29:如实施方案28所述的载体,其中所述抗镰状化人β-球蛋白基因包括约2.3kb的重组人β-球蛋白基因,所述重组人β-球蛋白基因包含在人β-球蛋白基因5'启动子和人β-球蛋白3'增强子控制下的外显子和内含子。
实施方案30:如实施方案29所述的载体,其中所述β-球蛋白基因包含来自ivs2的具有375bprsai缺失的β-球蛋白内含子2,以及包含hs2、hs3和hs4的复合人β-球蛋白基因座控制区。
实施方案31:一种病毒颗粒,其包含根据实施方案1至23中任一项所述的载体。
实施方案32:一种宿主细胞,其用根据实施方案2至23中任一项所述的载体转导。
实施方案33:根据实施方案32所述的宿主细胞,其中所述细胞是干细胞。
实施方案34:根据实施方案33所述的宿主细胞,其中所述细胞是源自骨髓的干细胞。
实施方案35:根据实施方案33所述的宿主细胞,其中所述细胞是并非源自胚胎或胚胎组织的干细胞。
实施方案36:根据实施方案32所述的宿主细胞,其中所述细胞是293t细胞。
实施方案37:根据实施方案32所述的宿主细胞,其中所述细胞是人造血祖细胞。
实施方案38:根据实施方案37所述的宿主细胞,其中所述人造血祖细胞是cd34+细胞。
实施方案39:根据实施方案37所述的宿主细胞,其中所述人造血祖细胞是cd34+/cd38-细胞。
实施方案40:一种用于治疗以下a列中所示的病变的组合物,所述组合物包含药学上可接受的载剂和用根据实施方案2至23中任一项所述的载体转染的干细胞和/或祖细胞,其中所述载体含有一种或多种如在以下b列中所示的用于治疗所述病变的转基因:
实施方案41:根据实施方案40所述的组合物,其中所述组合物用于治疗ada-scid,并且所述转基因表达腺苷脱氨酶(ada)。
实施方案42:根据实施方案40所述的组合物,其中所述组合物用于治疗x-scid,并且所述转基因表达il-2受体γ(il-2rγ)。
实施方案43:根据实施方案40所述的组合物,其中所述组合物用于治疗镰状细胞病,并且所述转基因表达抗镰状化人β-球蛋白基因。
实施方案44:如实施方案43所述的组合物,其中所述抗镰状化人β-球蛋白基因包括约2.3kb的重组人β-球蛋白基因,所述重组人β-球蛋白基因包含在人β-球蛋白基因5'启动子和人β-球蛋白3'增强子控制下的外显子和内含子。
实施方案45:如实施方案44所述的组合物,其中所述β-球蛋白基因包含来自ivs2的具有375bprsai缺失的β-球蛋白内含子2,以及包含hs2、hs3和hs4的复合人β-球蛋白基因座控制区。
实施方案46:根据实施方案40至45中任一项所述的组合物,其中所述宿主细胞是cd34+细胞。
实施方案47:根据实施方案46所述的组合物,其中所述宿主细胞是cd34+/cd38-细胞。
实施方案48:一种针对以下a列中所示的病变治疗受试者的方法,所述方法包括将用根据实施方案2至23中任一项所述的载体转染的祖细胞或干细胞引入所述受试者,其中所述载体含有一种或多种如在以下b列中所示的用于治疗所述病变的转基因:
实施方案49:根据实施方案48所述的方法,其中所述方法用于治疗ada-scid,并且所述转基因表达腺苷脱氨酶(ada)。
实施方案50:根据实施方案48所述的方法,其中所述方法用于治疗x-scid,并且所述转基因表达il-2受体γ(il-2rγ)。
实施方案51:根据实施方案48所述的方法,其中所述方法用于治疗镰状细胞病,并且所述转基因表达抗镰状化人β-球蛋白基因。
实施方案52:如实施方案51所述的方法,其中所述抗镰状化人β-球蛋白基因包括约2.3kb的重组人β-球蛋白基因,所述重组人β-球蛋白基因包含在人β-球蛋白基因5'启动子和人β-球蛋白3'增强子控制下的外显子和内含子。
实施方案53:如实施方案52所述的方法,其中所述β-球蛋白基因包含来自ivs2的具有375bprsai缺失的β-球蛋白内含子2,以及包含hs2、hs3和hs4的复合人β-球蛋白基因座控制区。
实施方案54:根据实施方案48至53中任一项所述的方法,其中所述引入包括用所述载体转导来自所述受试者的干细胞和/或祖细胞;以及将所述转导的细胞或源自所述转导的细胞的细胞移植到所述受试者中,其中所述细胞或来自所述细胞的衍生物表达所述转基因。
实施方案55:根据实施方案48至54中任一项所述的方法,其中所述细胞是祖细胞。
实施方案56:根据实施方案48至54中任一项所述的方法,其中所述细胞是干细胞。
实施方案57:根据实施方案48至56中任一项所述的方法,其中所述细胞来源于骨髓。
实施方案58:根据实施方案48至57中任一项所述的方法,其中所述细胞是cd34+细胞。
实施方案59:根据实施方案58所述的方法,其中所述细胞是cd34+/cd38-细胞。
实施方案60:根据实施方案48至59中任一项所述的方法,其中所述细胞来源于所述受试者。
实施方案61:一种提供改进的用重组慢病毒转导的细胞群体,所述细胞群体富含cd34+/cd38-细胞。
实施方案62:根据实施方案61所述的细胞群体,其中所述cd34+/cd38-细胞来源于血液或骨髓。
实施方案63:根据实施方案61至62中任一项所述的群体,其中所述cd34+/cd38-细胞用含有转基因的逆转录病毒载体转染。
实施方案64:根据实施方案63所述的细胞群体,其中所述cd34+/cd38-细胞用选自由以下各项组成的组的逆转录病毒载体转导:hiv-1慢病毒载体、hiv-2慢病毒载体、α逆转录病毒载体、马传染性贫血病毒(eiav)慢病毒载体、momlv载体、x-mlv载体、p-mlv载体、a-mlv载体、galv载体、hev-w载体、siv-1载体、fiv-1载体以及serv-1-5载体。
实施方案65:根据实施方案63所述的细胞群体,其中所述cd34+/cd38-细胞用慢病毒载体转导。
实施方案66:根据实施方案65所述的细胞群体,其中所述cd34+/cd38-细胞用非tat依赖性和自灭活(sin)慢病毒载体转导。
实施方案67:根据实施方案63至66中任一项所述的细胞群体,其中所述转基因是用于治疗在表1中列出的病变的转基因。
实施方案68:根据实施方案63至66中任一项所述的细胞群体,其中所述转基因编码ada、il-2γr或抗镰状化基因。
实施方案69:根据实施方案63所述的细胞群体,其中所述细胞用ccl-βas3-fblv转染。
实施方案70:一种改进干细胞或祖细胞的转导的方法,所述方法包括为所述转导提供富含cd34+/cd38-细胞的干细胞或祖细胞群体。
定义
“重组”与其在本领域中的使用一致地用于指包含某些部分的核酸序列,所述部分并非作为单一序列的一部分一起天然存在或相对于天然存在的序列已经重排。重组核酸通过涉及人工的方法产生和/或由通过人工产生的核酸产生(例如,通过复制、扩增、转录的一个或多个循环等)。重组病毒是包含重组核酸的病毒。重组细胞是包含重组核酸的细胞。
如本文所用,术语“重组慢病毒载体”或“重组lv”是指由于人干预和操作,由lv和多个另外区段组装的人工产生的多核苷酸载体。
“球蛋白核酸分子”是指编码球蛋白多肽的核酸分子。在各个实施方案中,所述球蛋白核酸分子可包含在编码序列的上游和/或下游的调控序列。
“球蛋白多肽”是指与人α、β或γ球蛋白具有至少85%、或至少90%、或至少95%、或至少98%氨基酸序列同一性的蛋白质。
术语“治疗性功能球蛋白基因”是指这样的核苷酸序列,所述核苷酸序列的表达产生球蛋白,所述球蛋白不产生血红蛋白病表型并且有效地向具有缺陷型球蛋白基因的个体提供治疗益处。所述功能性球蛋白基因可编码适用于待治疗的哺乳动物个体的野生型球蛋白,或者所述功能性球蛋白基因可以是球蛋白的突变形式,优选地是提供优异性质例如优异的氧转运性质或抗镰状化性质的球蛋白。所述功能性球蛋白基因包含外显子和内含子两者,以及球蛋白启动子和剪接供体/受体。
“有效量”是指所需药剂或包含所述药剂的组合物相对于未治疗的患者改善或消除疾病的症状的量。用于实践本文所述的用于治疗性治疗疾病的方法的组合物的有效量根据施用方式、受试者的年龄、体重和总体健康状况而变化。最终,主治医生或兽医将决定适当的量和给药方案。这种量被称为“有效”量。
附图简述
图1示意性地示出反向取向ubc慢病毒载体(pcclc-roubc)转移质粒图谱的一个实施方案。虚线指示慢病毒序列外的质粒骨架序列。
图2示出与改进的反向取向cclc-roubc-emgfp载体相比,自正向取向cclc-ubc-emgfp载体的表达。
图3示意性地示出用于研究的表达载体。慢病毒图描绘cclc载体中的位置。roubc和roubcs载体含有在emgfp阅读框的末端之后,处于适当反向取向的牛生长激素聚腺苷酸化信号(未描绘)。pcafe表达质粒含有在sv40聚腺苷酸化信号的上游的相同的盒。
图4,图a和b示出ubc剪接的基因分析。图a:pcr策略,示出引物位置和预期产物大小。图b:来自对照的pcr产物和来自用携带ubc启动子变体的慢病毒载体转导的细胞的gdna的电泳。
图5,图a-c示出在包装和转导期间ubc内含子丢失的定量分析。图a:用于定量标准化至总原病毒整合(hex-lvpsi)的ubc内含子拷贝(fam-ubc内含子)的双重ddpcr策略。图b:来自ddpcr的代表性原始数据,其说明正微滴与负微滴之间的分离。图c:ubc内含子拷贝与对照和用携带ubc启动子变体的lv转导的样品中的总原病毒拷贝的比率。误差线表示基于ddpcr泊松统计的95%置信区间。
图6,图a-c示出ubc启动子变体的流式细胞术表达分析。图a:用表达质粒转染后48小时,293t细胞的几何平均荧光强度(gmfi)。误差线表示三次生物重复实验的标准偏差。ubc对比ubcs非配对t检验p=0.0122,roubc对比roubcsp=0.0134。图b:用携带ubc启动子变体的cclc慢病毒载体转导后10天,k562细胞的gmfi。数据代表多次实验。图c:转染后第48小时,293t细胞的gmfi。误差线表示三次生物重复实验的sd。*表示p<0.05。ubc对比denh非配对t检验p=0.0267,ubcs对比denhp=0.0008。
图7,图a-e示出eef1a1分析。图a:携带eef1a1启动子变体的慢病毒载体的图。图b:在转导后大于2周,在稳定转导的k562细胞中在整个eef1a1内含子上扩增的pcr产物的凝胶电泳。图c:在(图b)中分析的样品中内含子拷贝与原病毒拷贝的比率的ddpcr定量。图d:通过流式细胞术测量的用表达质粒转染后48小时,瞬时转染的293t细胞的gmfi。图c中的误差线表示95%置信区间,并且在图d和e中表示三次生物重复实验的sd。*表示p<0.05。非配对t检验p=0.0064。(e)通过流式细胞术测量的转导后10天,稳定转导的k562细胞的gmfi。
图8图a-c示出双向载体分析。图a:载体示意图。图b:bd和robd载体中内含子丢失的ddpcr分析。图c:通过流式细胞术测量的转导后2周,稳定转导的293t细胞的gmfi。误差线表示95%置信区间。
图9示出病毒载体上清液和转导的k562细胞中剪接的载体接点(junction)的数字pcr定量。
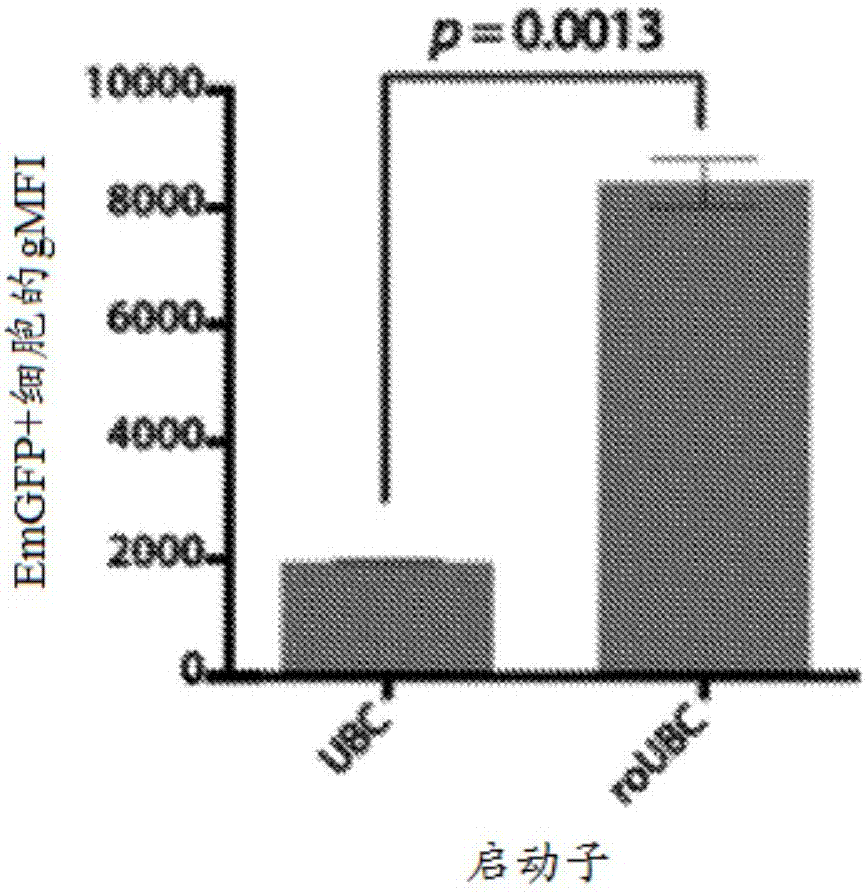
图10示出通过流式细胞术测量的骨髓细胞中的emgfp的表达,所述骨髓细胞分化自从健康供体的流动的外周血液富集的转导的人cd34+hspc。在转导后10天分析约10%转导的群体中的表达。双尾t检验p值0.0013。
图11示出在转导后10天,在转导的cd34+hspc中呈原病毒形式的ubc内含子的数字pcr定量。
图12示出在基于pgl4.25的质粒中,内含子序列的增强子活性的荧光素酶测定的结果。在转染293t细胞后48小时,测量细胞溶解产物中的荧光素酶活性。
图13,图a-c示出内含子交换的ubc和eef1a1慢病毒载体的表达和基因分析。图a:在转导后7天通过流式细胞术测量的用含有所指示启动子的慢病毒载体转导的k562细胞的表达分析。图b:使用图4,图a中所图示的引物进行的基因分析。在右侧的三个泳道中中等长度的产物是来自k562基因组dna的非特异性产物且应被忽略。图c:使用图7,图a中所图示的引物对转导的k562细胞的基因分析。
图14,图a-b示出人cd34+和cd34+/cd38-细胞的分离和生长特性。图a:富含cd34-的细胞的流式细胞术,示出用于限定cd34+/cd38+细胞(区域p5)和cd34+/cd38-细胞(区域p3)的设门策略。图b:从来自脐带血的cd34+和cd34+/cd38-细胞的细胞扩增。将细胞与照射的ms5基质细胞在长期培养基中共培养。在长期培养的每个时间点示出相对于在第0天接种的细胞数目的平均倍数增加。数据表示随时间推移的细胞扩增±sem(n=3,p<.0001)。缩写:apc,别藻蓝蛋白。
图15,图a-f示出用ccl-βas3-fblv载体转导cd34+和cd34+/cd38-细胞的分析。图a:转导的cd34+和cd34+/cd38-细胞中的载体拷贝数目(vcn)±sem(n=9,p=.02)。图b:由非转导的脐带血(cb)cd34+(nt-cd34+)细胞、转导的cd34+(cd34+)细胞和cd34+/cd38-细胞形成的造血集落类型(n=80个集落)的分布。图c:在体外生长成造血集落的接种的nt-cd34+细胞、cd34+细胞和cd34+/cd38-细胞的百分比。值表示平均值±sd。图d:通过ddpcr分析由转导的cd34+(左)细胞和cd34+/cd38-(右)cb细胞生长的单cfu的vcn(n=80个集落)。图表指示通过数字pcr对于载体为阴性(0个vc/细胞)或者具有1-2、3-4、5-6或>6个vc/细胞的cfu的百分比。图e:cd34+细胞和cd34+/cd38-细胞的载体转导剂量反应(n=3,在6.6x106tu/ml下p=.05,在2x107tu/ml下p=.002)。图f:在长期培养中随时间推移的vcn(±sem[n=3])(时间趋势差p=.03,vcn差异p=.004,线性混合模型)。星号指示显著性,*,p≤.05;**,p≤.01。
图16,图a-c示出ccl-mnd-gfplv载体对cd34+细胞和cd34+/cd38-细胞的转导的分析。图a:在培养的第14天通过qpcr分析,在用一定剂量范围的ccl-mnd-gfplv转导后平均载体拷贝数目±sem的比较。图b:示出转导的cd34+细胞和cd34+/cd38-细胞的相对gfp表达的代表性直方图。图c:在ccl-mnd-gfp转导的cd34+细胞和cd34+/cd38-细胞中通过流式细胞术测定的gfp1细胞的百分比(n=6,p=.02)。缩写:gfp,绿色荧光蛋白。
图17,图a-b示出通过ccl-βas3-fblv载体转导的cd34+细胞和cd34+/cd38-细胞的红系细胞分化。图a:在转导后第14天,在分化期间的载体拷贝数目(vcn)±sem的比较(n=3)。图b:通过qrt-pcr分析的从转导的cd34+细胞和cd34+/cd38-细胞分化的红系细胞中所有β-球蛋白转录物/vcn的hbbas3mrna表达的百分比(%as3/vcn)(n=3)。
图18,图a-e示出载体包膜和受体对由ccl-βas3-fblv载体转导的作用。在以下各项上cd34+和cd34+/cd38-细胞的ldl受体表达:(图a)新鲜分离的cd34+细胞,(图b)在细胞因子中培养48小时的cd34+细胞,(图c)新鲜分离的cd34+/cd38-细胞,和(图d)在细胞因子中培养48小时的cd34+/cd38-细胞。图e:用rd114假型ccl-βas3-fblv载体转导cd34+和cd34+/cd38-细胞。所述图表示在培养的第14天通过qpcr分析的cd34+细胞和cd34+/cd38-细胞的平均载体拷贝数目±sem(n=3)。缩写:ldl,低密度脂蛋白。
图19,图a和b示出nod.cg-prkdscidil2rgtm1wjil/szj(nsg)小鼠植入的比较。图a:转导的移植细胞群体对nsg小鼠中的人cd451细胞植入的贡献。对mock小鼠移植未转导的人cbcd34+细胞;对对照小鼠移植转导的cd34+细胞;对所有其他小鼠移植cd34+/cd38-细胞(1%)和cd34+/cd38+细胞(99%)的组合。将用于转导的载体(cclc-ubc-mstrawberry-fb,cclc-ubc-mcitrine-fb,和cclc-ubcmcerulean-fblv)在用于每个移植的细胞群体之间交替。使用流式细胞术进一步分析从具有人细胞植入的nsg小鼠收获的bm(%hucd451/%hucd4511mucd451细胞)的载体表达百分比。(b):在体内小鼠移植中分析的细胞的载体拷贝数目(vcn)。用对每个荧光报告分子具有特异性的引物和探针使用ddpcr,从移植到nsg小鼠中之后80-90天收获的bm分析体内vcn。分别展示每个细胞群体的体内vcn/小鼠。
图20,图a-c示出在长期培养的第30天,对用ccl-βas3-fblv载体的cd34+和cd34+/cd38-细胞转导和造血潜能的分析。图a:通过非转导的(nt)-cd34+细胞(n=37个集落)、转导的cd34+细胞(n=29个集落)和cd34+/cd38-细胞(n=81个集落)形成的造血集落类型的分布。图b:在体外生长成造血集落的接种的nt-cd34+细胞、转导的cd34+细胞和cd34+/cd38-细胞的百分比。值表示平均值±sd;星号指示显著性,****p≤0.0001。图c:通过ddpcr分析的从转导的cd34+生长的体外单一cfu的vcn分布(n=22个集落)。图指示对于载体为阴性(0个vc/细胞)或者具有1-2、3-4、5-6或>6个vc/细胞的cfu的百分比。从转导的cd34+/cd38-细胞生长的体外cfu的vcn分布(n=43个集落)。
图21,图a-c示出在长期培养的第60天,对用ccl-βas3-fblv载体的cd34+和cd34+/cd38-细胞转导和造血潜能的分析。图a:通过非转导的(nt)-cd34+细胞(n=5个集落)、转导的cd34+细胞(n=3个集落)和cd34+/cd38-细胞(n=22个集落)形成的造血集落类型的分布。图b:在体外生长成造血集落的接种的nt-cd34+细胞、转导的cd34+细胞和cd34+/cd38-细胞的百分比。图c:通过ddpcr分析的从转导的cd34+/cd38-细胞生长的体外单一cfu的vcn分布(n=18个集落)。图指示对于载体为阴性(0个vc/细胞)或者具有1-2、3-4、5-6或>6个vc/细胞的cfu的百分比。
图22,图a-c示出通过ccl-βas3-fblv载体转导的cd34+细胞和cd34+/cd38-细胞的红系细胞分化。对来自(图a)未分级分离的cd34+细胞和(图b)cd34+/cd38-细胞的红系细胞培养物的细胞的流式细胞术分析。无核红细胞在左上象限中呈现为draq5阴性血型糖蛋白a(gpa)阳性细胞。图c:在红系细胞分化结束时无核rbc的百分比。
图23示出转导的移植细胞群体对nsg小鼠中的总人植入的贡献。对mock小鼠移植未转导的人cbcd34+细胞;对对照小鼠移植转导的cd34+细胞;对所有其他小鼠移植cd34+/cd38-细胞(1%)和cd34+/cd38+细胞(99%)的组合。将用于转导的载体(cclc-ubc-mstrawberry-fb,cclc-ubc-mcitrine-fb和cclc-ubc-mcerulean-fblv)在用于每个移植的细胞群体之间交替。使用流式细胞术进一步分析从具有人细胞植入的nsg小鼠收获的bm(%hucd45+/%hucd45++mucd45+细胞)的载体表达百分比。
详述
基于hiv-1的慢病毒载体(lv)是在生物学实验和基因疗法中用于遗传修饰的最常用工具之一。所使用的大多数lv是自灭活的,意味着含有启动子和增强子的长末端重复序列内的区域已被除去(zufferey等人(1998)j.virol.,72:9873-9880)。为了在这种载体内表达转基因,因此必须将启动子与转基因一起置于载体有效负载内。通常,为了表达编码蛋白质的基因,将使用异源rnapolii病毒或细胞启动子,并且常见的实例是来自巨细胞病毒、鼠白血病病毒和脾病灶形成病毒的病毒启动子,以及来自人基因的细胞启动子,如延伸因子1α(eef1a1)、泛素c(ubc)和磷酸甘油酸激酶(pgk1)(schambach等人(2006)mol.ther.,13,391-400;dull等人(1998)j.virol.,72:8463-8471)。
在病毒产生过程中,rnapolii转录载体基因组,通常从已转染到生产细胞中的转移质粒中转录。几乎所有系统都并入来自hiv-1的rev蛋白,所述rev蛋白与hiv-1基因组内的rev响应元件(rre)结合并介导病毒基因组的非剪接依赖性核输出。尽管将rre序列并入lv构建体中,但是如果剪接事件在转录物中保留包装信号(psi),则所述载体有效负载内的内含子可在包装期间丢失。但是,对于一些表达盒,如包含含内含子的eef1a1启动子的表达盒和含有杂合cag启动子的表达盒,在慢病毒包装期间尚未观察到内含子丢失(ramezani等人(2000)mol.ther.,2:458-469;zaiss等人(2002)j.virol.,76:7209-7219)。根据这些观察结果,有时推断慢病毒基因转移允许内含子的传递(logan等人(2002)curr.opin.biotechnol.,13:429-436)。
着手调查人ubc启动子所含的内含子是否由转移质粒忠实地传递至稳定转导的细胞中的原病毒形式。假设ubc内含子的丢失将导致转基因表达的显著降低,因为据报道ubc内含子具有强烈的增强子活性(bianchi等人(2009)gene,448:88-101)。与eef1a1内含子的先前发现相比,发现ubc内含子在用从含内含子的质粒产生的载体转导的细胞中的大部分原病毒形式中缺失。ubc内含子的缺乏导致细胞系中瞬时转染实验和稳定转导实验两者中的表达减少约2倍,以及原代细胞中的转导实验中减少4倍。这与使用eef1a1启动子的实验形成显著对比,在使用eef1a1启动子的实验中大部分原病毒形式保留所述内含子。ubc表达盒的逆转阻止了这种剪接介导的内含子丢失和在单向和双向lv中的最大化表达。不受特定理论的束缚,据信ubc与eef1a1启动子之间的内含子维持的差异由启动子外显子序列而不是内含子序列本身造成。
鉴于前述,在各个实施方案中,提供了包含人泛素c(ubc)启动子的重组逆转录病毒载体,其中所述ubc启动子在所述载体中处于反向取向,以使得从所述启动子转录的方向朝向所述载体的5'长末端重复序列(ltr)定向。在各个实施方案中,所述载体包含多克隆位点,所述多克隆位点被定位成使得插入所述多克隆位点中的基因/cdna与反向取向ubc可操作地连接,以使得受所述启动子控制的基因的转录方向朝向所述载体的5'长末端重复序列(ltr)定向。
作为非限制性说明,图1中示出一种这样的病毒载体。为了制备这种慢病毒载体“pcclc-roubc”,将来自人泛素c基因的片段(ucsc人基因组序列型式hg19,减去125398318位至125399530位的链)使用标准分子克隆技术(如限制性消化和连接)或组装技术in-fusion、gibson组装或不依赖于序列和连接的克隆(slic)插入pcclc的多克隆位点中。
所述插入方向是使得自ubc启动子转录的方向朝向pcclc载体的5'长末端重复序列(ltr)定向,这与具有朝向3'ltr定向的转录方向的典型慢病毒载体不同。
在靶细胞的慢病毒转导后,与具有定向为使得转录朝向3'ltr进行的ubc启动子的载体相比,这种roubc载体以大约高四倍的水平表达转基因。所述值是在用编码绿色荧光蛋白(emgfp)转基因的emerald变体的载体转导的人造血干细胞和祖细胞中测定的(图2)。
这种启动子取向的使用不限于pcclc慢病毒载体,并且据信在其他逆转录病毒载体中也是有益的。因此,在某些实施方案中,考虑了包含反向取向ubc启动子的其他逆转录病毒载体。此类载体包括但不限于hiv-2慢病毒载体、α逆转录病毒载体、马传染性贫血病毒(eiav)慢病毒载体、momlv载体、x-mlv载体、p-mlv载体、a-mlv载体、galv载体、hev-w载体、siv-1载体、fiv-1载体、serv-1-5载体等。
在各个实施方案中,所述载体另外包含以相同(反向)取向插入启动子片段的3'(相对于整个载体序列启动子的5')中的聚腺苷酸化信号(polya),以便实现转基因的有效聚腺苷酸化。合适的聚腺苷酸化信号包括但不限于牛生长激素polya、人生长激素polya、兔β-球蛋白基因聚腺苷酸化信号、人疱疹病毒(hsv)聚腺苷酸化信号、胸苷激酶(tk)基因聚腺苷酸化信号等。
还发现ubc启动子的逆转还改进来自双向载体的表达,如在美国专利号8,501,464b2中描述的双向载体。这通过与正向取向对应物“bd”相比,具有反向ubc启动子“robd”的双向载体中的egfp的表达增加来证明(参见例如图8,图c)。因此,在某些实施方案中,还考虑了包含处于反向取向的人ubc基因的双向逆转录病毒载体(例如,双向慢病毒载体)。
在各个实施方案中,还考虑了包含本文所述的载体的病毒颗粒以及用本文所述的载体转导的宿主细胞(例如,干细胞、祖细胞等)。在某些实施方案中,所述宿主细胞是cd34+造血干细胞。如本文在实施例2中所描述,发现使用分离的cd34+/cd38-允许使用显著较少的载体并且似乎改进用于hsc基因疗法的转导。因此,在某些实施方案中,所述宿主细胞是cd34+/cd38-细胞,并且在某些实施方案中,提供了富含cd34+/cd38-细胞的细胞群体。
cd34+/cd38-细胞的使用不必限于用含有反向取向ubc的载体转导。在某些实施方案中,可用基本上任何逆转录病毒载体(例如,在pct公布号:wo2014043131a1(pct/us2013/059073)中所描述的抗镰状化逆转录病毒载体,如ccl-βas3-fblv)来转导此类细胞。
在各个实施方案中,提供了用于治疗病变的组合物,其中所述组合物包含用如本文所述的载体转染的干细胞和/或祖细胞,其中所述载体含有一种或多种用于治疗病变的转基因(例如,如以下表1中所示),其中所述组合物另外包含药学上可接受的载剂。
在某些实施方案中,考虑了治疗病变(例如,可通过引入转基因治疗的病变的方法。在某些实施方案中,所述方法包括将用本文所述的载体转染的祖细胞或干细胞引入患有所述病变或处于所述病变风险的受试者,其中所述载体含有一种或多种用于治疗所述病变的转基因(例如,如以下表1中所示)。
应注意,尽管下文提供的论述是关于sin慢病毒载体,但使用本文提供的各种元件、构建体和教义,可容易地产生含有反向取向ubc启动子或包含反向取向ubc启动子的双向启动子的其他逆转录病毒载体。
非tat依赖性和自灭活慢病毒载体.
如上所述,设想反向取向ubc启动子可用于基本上任何逆转录病毒载体中。在某些实施方案中,所述逆转录病毒载体是慢病毒载体(lv),并且在某些实施方案中,所述慢病毒载体(lv)包含非tat依赖性和自灭活(sin)构型。因此,在各个实施方案中,希望在本文所述的lv中使用相对于野生型ltr具有降低的启动子活性的ltr区域。可提供此类构建体,所述构建体实际上是“自灭活的”(sin),其提供生物安全特征。sin载体是其中转导细胞中全长载体rna的产生大大减少或完全消除的载体。这种特征使有复制能力的重组体(rcr)将出现的风险最小化。此外,它降低了位于载体整合位点附近的细胞编码序列将被异常表达的风险。
此外,sin设计降低了ltr与驱动转基因表达的启动子之间的干扰的可能性。sinlv通常可允许内部启动子的完全活性。
sin设计增加了lv的生物安全性。hivltr的大部分由u3序列组成。u3区含有增强子和启动子元件,所述增强子和启动子元件在感染的细胞中并响应于细胞活化调节hiv基因组的基础和诱导性表达。这些启动子元件中的几种对于病毒复制是必需的。所述增强子元件中的一些在病毒分离株中是高度保守的,并且一直牵涉于病毒发病机制中的关键毒力因子。增强子元件可用于影响病毒的不同细胞靶标中的复制速率。
由于病毒转录开始于5'ltr的u3区的3'末端,所以那些序列不是病毒mrna的一部分,并且来自3'ltr的其拷贝充当用于在整合的原病毒中产生两种ltr的模板。如果u3区的3'拷贝在逆转录病毒载体构建体中被改变,则载体rna仍然由生产细胞中的完整5'ltr产生,但不能在靶细胞中再生。这种载体的转导导致子代病毒中两种ltr的灭活。因此,逆转录病毒是自灭活的(sin),并且那些载体被称为sin转移载体。
在某些实施方案中,通过在载体dna的3'ltr的u3区(即用于产生载体rna的dna)中引入缺失来实现自灭活。在rt期间,这种缺失被转移至原病毒dna的5'ltr。通常,希望消除足够的u3序列以极大地减少或完全消除ltr的转录活性,从而极大地减少或消除转导细胞中全长载体rna的产生。然而,通常希望保留参与病毒rna的聚腺苷酸化(通常散布在u3、r和u5上的功能)的ltr的那些元件。因此,在某些实施方案中,希望尽可能多地从ltr消除转录上重要的基序,同时省去聚腺苷酸化决定簇。
sin设计在zufferey等人(1998)jvirol.72(12):9873-9880和美国专利号:5,994,136中详细描述。如其中所描述,然而,存在对3'ltr处的缺失程度的限制。首先,u3区的5'末端在载体转移中起到另一种基本功能,所述功能是整合所需的(末端二核苷酸+att序列)。因此,末端二核苷酸和att序列可表示可缺失的u3序列的5'边界。另外,一些宽松限定的区域可影响r区中的下游聚腺苷酸化位点的活性。来自3'ltr的u3序列的过度缺失可减少载体转录物的聚腺苷酸化,对生产细胞中的载体的滴度和靶细胞中的转基因表达两者均具有不利影响。
在美国专利公布号:2003/0039636中描述了另外的sin设计。如其中所描述,在某些实施方案中,从ltr中除去的慢病毒序列被来自非慢病毒逆转录病毒的相当序列置换,从而形成杂合ltr。具体地说,ltr内的慢病毒r区可全部或部分地被来自非慢病毒逆转录病毒的r区置换。在某些实施方案中,慢病毒tar序列(与tat蛋白相互作用以增强病毒复制的序列)优选整个从r区除去。然后用来自非慢病毒逆转录病毒的r区的相当部分置换tar序列,从而形成杂合r区。可进一步修饰ltr以除去慢病毒u3和u5区的全部或一部分和/或用非慢病毒序列置换慢病毒u3和u5区的全部或一部分。
因此,在某些实施方案中,sin构型提供包含缺乏其tar序列的全部或一部分的杂合慢病毒r区的逆转录病毒ltr,从而消除任何可能的由tat引起的活化,其中所述tar序列或其部分被来自非慢病毒逆转录病毒的r区的相当部分置换,由此形成杂合r区。在一个具体实施方案中,所述逆转录病毒ltr包含杂合r区,其中所述杂合r区包含hivr区的缺乏tar序列的一部分(例如,包含在us2003/0039636中的seqidno:10中所示的核苷酸序列或由所述核苷酸序列组成的一部分)以及momsvr区的与从hivr区缺乏的tar序列相当的一部分(例如,包含在2003/0039636中的seqidno:9中所示的核苷酸序列或由所述核苷酸序列组成的一部分)。在另一个具体实施方案中,整个杂合r区包含在2003/0039636中的seqidno:11中所示的核苷酸序列或由所述核苷酸序列组成。
r区可源自其的合适慢病毒包括例如hiv(hiv-1和hiv-2)、eiv、siv和fiv。非慢病毒序列可源自其的合适逆转录病毒包括例如momsv、momlv、弗兰德(friend)、mscv、rsv和泡沫病毒。在一个说明性实施方案中,慢病毒是hiv,并且非慢病毒逆转录病毒是momsv。
在us2003/0039636中描述的另一个实施方案中,包含杂合r区的ltr是左侧(5')ltr并且还包含在杂合r区上游的启动子序列。优选的启动子起源于非慢病毒,并且包括例如来自非慢病毒逆转录病毒的u3区(例如,momsvu3区)。在一个具体实施方案中,所述u3区包含在us2003/0039636中的seqidno:12中所示的核苷酸序列。在另一个实施方案中,左侧(5')ltr还包含在杂合r区下游的慢病毒u5区。在一个实施方案中,所述u5区是hivu5区,包括基因组整合所需的hivatt位点。在另一个实施方案中,所述u5区包含在us2003/0039636中的seqidno:13中所示的核苷酸序列。在又一个实施方案中,整个左侧(5')杂合ltr包含在us2003/0039636中的seqidno:1中所示的核苷酸序列。
在另一个说明性实施方案中,包含杂合r区的ltr是右侧(3')ltr并且还包含在杂合r区上游的修饰的(例如,截短的)慢病毒u3区。修饰的慢病毒u3区可包含att序列,但缺乏具有启动子活性的任何序列,从而使得载体是sin,因为病毒转录不能超越染色体整合后的第一轮复制。在一个具体实施方案中,在所述杂合r区上游的修饰的慢病毒u3区由慢病毒(例如,hiv)u3区的3'端直至慢病毒u3att位点(且包括慢病毒u3att位点)组成。在一个实施方案中,所述u3区包含在us2003/0039636中的seqidno:15中所示的核苷酸序列。在另一个实施方案中,右侧(3')ltr还包含在杂合r区下游的聚腺苷酸化序列。在另一个实施方案中,所述聚腺苷酸化序列包含在us2003/0039636中的seqidno:16中所示的核苷酸序列。在又一个实施方案中,整个右侧(5')ltr包含在us2003/0039636的seqidno:2或17中所示的核苷酸序列。
因此,在基于hiv的lv的情况下,已经发现此类载体耐受显著u3缺失,包括除去ltrtata盒(例如,缺失-418至-18),而无载体滴度的显著降低。这些缺失使得ltr区基本上无转录活性,因为ltr的转录能力降低至约90%或更低。
还已经证明,如果转移载体构建体中的上游ltr的一部分被组成型活性启动子序列置换,则tat的反式作用功能变得可有可无(参见例如,dull等人(1998)jvirol.72(11):8463–8471。此外,表明处于反式的rev的表达允许从仅含有gag和pol的包装构建体产生高滴度的hiv源性的载体原料(vectorstocks)。这种设计使包装功能的表达取决于仅在生产细胞中可获得的互补。所得到的基因递送系统仅保留hiv-1的九种基因中的仅三种,并且依赖于四个单独的转录单元来产生转导颗粒。
在实施例1中所示的一个实施方案中,表达抗镰状化β-球蛋白(例如,βas3)的盒被置于pccllv骨架中,其是cmv增强子/启动子在5'ltr中被取代的sin载体。
应认识到,cmv启动子通常提供高水平的非组织特异性表达。具有相似组成型活性的其他启动子包括但不限于rsv启动子和sv40启动子。也可使用哺乳动物启动子,如β-肌动蛋白启动子、泛素c启动子、延伸因子1α启动子、微管蛋白启动子等。
前述sin构型是说明性的而非限制性的。许多sin构型是本领域的技术人员已知的。如上文所指示,在某些实施方案中,ltr转录被减少约95%至约99%。在某些实施方案中,可使ltr至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%或至少约99%无转录活性。
绝缘子元件
在某些实施方案中,为了进一步增强生物安全性,将绝缘子插入本文所述的载体中。绝缘子是整个基因组中存在的dna序列元件。它们结合修饰染色质并改变区域基因表达的蛋白质。本文描述的载体中的绝缘子的放置提供了各种潜在益处,尤其包括:1)通过侧翼染色体来保护载体免于表达的位置效应花斑(positionaleffectvariegation)(即屏障活性);以及2)保护侧翼染色体免于所述载体的基因表达的插入反式活化(增强子阻断)。因此,绝缘子可有助于保持嵌入基因组或基因背景中的基因或转录单元的独立功能,在所述基因组或基因背景中所述绝缘子的表达可另外受所述基因组或遗传背景内的调控信号影响(参见例如,burgess-beusse等人(2002)proc.natl.acad.sci.usa,99:16433;和zhan等人(2001)hum.genet.,109:471)。在本发明背景下,绝缘子可有助于保护慢病毒表达的序列免受整合位点效应,所述整合位点效应可由存在于基因组dna中的顺式作用元件介导并导致转移序列的表达失调。在各个实施方案中,提供lv,其中将绝缘子序列插入整合到细胞基因组中的载体区域中的一个或两个ltr或其他地方中。
第一且最佳表征的脊椎动物染色质绝缘子位于鸡β-球蛋白基因座控制区内。含有dna酶-i超敏位点-4(chs4)的此元件似乎构成鸡β-球蛋白基因座的5'边界(prioleau等人(1999)emboj.18:4035-4048)。含有chs4元件的1.2-kb片段展示典型绝缘子活性,包括阻断细胞系中球蛋白基因启动子和增强子的相互作用的能力(chung等人(1993)cell,74:505-514),以及保护果蝇(同上)、转化的细胞系(pikaart等人(1998)genesdev.12:2852-2862)和转基因哺乳动物(wang等人(1997)nat.biotechnol.,15:239-243;taboit-dameron等人(1999)transgenicres.,8:223-235)中的表达盒免于位置效应。大部分这种活性都包含在250-bp片段中。在此段内是49-bpchs4核心(chung等人(1997)proc.natl.acad.sci.,usa,94:575-580),其与牵涉于增强子阻断测定中的锌指dna结合蛋白ctcf相互作用(bell等人(1999)cell,98:387-396)。
一种说明性和合适的绝缘子是fb(fii/bead-a)(一种77bp绝缘子元件),其含有鸡β-球蛋白5'hs4绝缘子的最小ctcf结合位点增强子阻断组分和来自由ramezani等人(2008)stemcell26:3257-3266描述的人t细胞受体α/δ阻断元件α/δi(bead-1)绝缘子的同源区。fb“合成”绝缘子具有完全增强子阻断活性。这种绝缘子是说明性而非限制性的。可使用其他合适的绝缘子,包括例如全长鸡β-球蛋白hs4或其绝缘子子片段、锚蛋白基因绝缘子和其他合成绝缘子元件。
包装信号。
在各个实施方案中,本文所述的载体还包含包装信号。“包装信号”、“包装序列”或“psi序列”是足以指导序列包含包装信号的核酸包装到逆转录病毒颗粒中的任何核酸序列。所述术语包括天然存在的包装序列以及还有其工程改造的变体。包括慢病毒在内的多种不同的逆转录病毒的包装信号在本领域中是已知的。
rev响应元件(rre)。
在某些实施方案中,本文描述的载体包含rev响应元件(rre)以增强未剪接rna的核输出。rre是本领域的技术人员所熟知的。说明性rre包括但不限于rre,如位于hivnl4-3基因组(genbank登录号af003887)中的位置7622-8459处的rre以及来自hiv或逆转录病毒的其他菌株的rre。此类序列可从genbank或从数据库以urlhiv-web.lanl.gov/content/index容易地获得。
中央聚嘌呤段(cppt)。
在各个实施方案中,本文所述的载体还包含中央聚嘌呤段。例如已知在慢病毒(例如,hiv-1)载体构建体中插入含有中央聚嘌呤段(cppt)的片段据报道通过促进病毒cdna通过中心dnaflap的核输入而显著增强转导效率。
刺激表达的转录后调控元件(pre)
在某些实施方案中,本文所述的载体可包含多种转录后调控元件(pre)中的任一种,所述转录后调控元件在转录物中的存在增加蛋白质水平下的异源核酸(例如,ada、il-2rγ、βas3等)的表达水平。pre可特别适用于某些实施方案中,尤其是涉及具有适度启动子的慢病毒构建体的那些实施方案。
一种类型的pre是位于表达盒内的能够刺激基因表达的内含子。然而,在慢病毒的生命周期事件中,内含子可被剪接出去。因此,如果内含子用作pre,则通常将其置于与载体基因组转录物相反的取向。
不依赖于剪接事件的转录后调控元件提供了在病毒生命周期期间不被除去的优点。一些实例是单纯疱疹病毒的转录后加工元件,乙型肝炎病毒(hpre)和土拨鼠肝炎病毒(wpre)的转录后调控元件。在这些之中wpre通常是优选的,因为它包含未在hpre中发现的另外的顺式作用元件。这种调控元件通常位于载体内,以便包含在转基因的rna转录物中,但在转基因翻译单位的终止密码子之外。
wpre在美国专利号6,136,597中进行了表征和描述。如其中所描述,wpre是介导rna从细胞核至细胞质的有效转运的rna输出元件。它通过插入顺式作用核酸序列来增强转基因的表达,以使得元件和转基因被包含在单个转录物内。wpre在有义取向上的存在显示使转基因表达增加达7至10倍。逆转录病毒载体转移呈cdna而不是含完整内含子的基因形式的序列,因为内含子通常在引起形成逆转录病毒颗粒的事件序列期间被剪接出去。内含子介导初级转录物与剪接机构(splicingmachinery)的相互作用。因为剪接机构对rna的加工有助于其胞质输出,由于剪接机构与转运机构之间的结合,通常无效地表达cdna。因此,将wpre包含在载体中产生增强的转基因表达。
转导的宿主细胞和细胞转导的方法。
本文描述的重组载体和所得病毒能够将核酸序列(例如,编码抗镰状化β-球蛋白、ada、il-2rγ基因、表1中列出的任何其他靶标/转基因等的核酸)转移到哺乳动物细胞中。为了递送至细胞,本发明的载体可与合适的包装细胞系结合使用,或在体外与含有必需逆转录病毒基因(例如,gag和pol)的其他载体质粒共同转染到细胞中以形成能够包装本发明的载体并感染细胞的无能的无复制能力的病毒粒子。
通常,所述载体经由转染引入包装细胞系中。包装细胞系产生含有载体基因组的病毒颗粒。转染方法是本领域的技术人员所熟知的。在将包装载体和转移载体共转染到包装细胞系中后,从培养基中回收重组病毒并通过本领域的技术人员使用的标准方法进行滴定。因此,可通过磷酸钙转染、脂质转染或电穿孔来将包装构建体(通常与显性选择性标记如新霉素、dhfr、谷氨酰胺合成酶一起)引入人细胞系中,随后在适当药物存在下选择并分离克隆。在某些实施方案中,选择性标记基因可与构建体中的包装基因物理地连接。
包装功能被配置为由合适的包装细胞表达的稳定细胞系是已知的(参见例如描述包装细胞的美国专利号5,686,279)。一般来说,为了产生病毒颗粒,可使用与慢病毒gag和pol基因的表达相容的任何细胞,或者可被工程改造以支持这种表达的任何细胞。例如,可使用生产细胞,如293t细胞和ht1080细胞。
在其中并入逆转录病毒载体(例如慢病毒载体)的包装细胞形成生产细胞。因此,生产细胞是能够产生或释放携带目标治疗性基因(例如抗镰状化β-球蛋白、ada、il-2rγ基因等)的包装的感染性病毒颗粒的细胞或细胞系。这些细胞可进一步是锚定依赖性的,这意味着当这些细胞附着至诸如玻璃或塑料的表面上时,这些细胞将最佳地生长、存活或维持功能。当载体具有复制能力时,用作慢病毒载体包装细胞系的锚定依赖性细胞系的一些实例是hela或293细胞和perc.6细胞。
因此,在某些实施方案中,提供了将基因递送至细胞的方法,所述基因然后被整合到所述细胞的基因组中,所述方法包括使所述细胞与含有本文所述的慢病毒载体的病毒粒子接触。细胞(例如呈组织或器官的形式)可与病毒粒子离体接触(例如感染)且然后递送至将表达所述基因(例如,抗镰状化β-球蛋白、ada、il-2rγ基因等)的受试者(例如,哺乳动物、动物或人)。在各个实施方案中,所述细胞可以是与受试者自体同源的(即来自所述受试者),或者其可以是与受试者非自体同源的(即,同种异体的或异种的)。此外,因为本文描述的载体能够被递送至分裂细胞和非分裂细胞两者,所以所述细胞可来自各种各样,包括例如骨髓细胞、间充质干细胞(例如,获自脂肪组织)以及源自人和动物来源的其他原代细胞。或者,病毒粒子可直接体内施用至受试者或受试者的局部区域(例如骨髓)。
当然,如上所述,本文描述的载体特别适用于转导从骨髓、外周血或脐带血获得的人造血祖细胞或造血干细胞,以及转导cd4+t细胞、外周血b淋巴细胞或t淋巴细胞等。在某些实施方案中,特别优选的靶标是cd34+细胞。在某些实施方案中,所述靶标是cd34+/cd38-细胞。
基因疗法。
在某些实施方案中,本文所述的载体适用于将转基因引入受试者,例如以治疗可通过校正遗传缺陷和/或通过表达一种或多种异源基因而改善的病变。在一个说明性、但非限制性的实施方案中,所述方法包括使包含造血干细胞的人细胞群体与本文所述的包含目标转基因的载体在实现由所述载体转导所述群体中的人干细胞或祖细胞的条件下接触。所述干细胞可取决于最终的应用而在体内或体外转导。即使在人基因疗法(如人干细胞的基因疗法)的背景下,也可在体内转导所述干细胞或祖细胞,或者在体外转导、然后将转导的细胞注入人受试者中。在一方面,人细胞可使用本领域的技术人员熟知的方法从人(例如人患者)中移除,并如上所述进行转导。然后将转导的细胞重新引入同一或不同的人中,其中所述转基因的表达改善病变的一种或多种症状,或有效地治愈病变,或减缓病变的进展。
病变和基因疗法的靶标
本文所述的载体适用于递送转基因以治疗基本上可使用基因疗法技术治疗的任何病状。可用于递送转基因以治疗许多病变。在此方面,注意到大量基因治疗临床方案被批准或审查(参见例如,misra(2013)j.a.p.i.,61:127-133等)。
在某些实施方案中,载体含有用于治疗病变如scid、镰状细胞病、脂质体贮积症、囊性纤维化、肌营养不良、苯丙酮尿症、帕金森氏病或血友病的转基因。表1中示出了可使用本文描述的载体用基因转移(例如,基因疗法)方法治疗的病变和相关“靶标”的说明性但非限制性列表。
表1.可通过基因疗法治疗的病变和相关靶标/基因产物的说明性但非限制性实例。
在一个说明性实施方案中,本文所述的载体用于通过引入腺苷脱氨酶(ada)基因/cdna来治疗ada-scid。在另一个实施方案中,本文所述的载体用于通过引入il-2受体γ(il-2γ)基因来治疗x-scid。在某些实施方案中,本文所述的载体适用于通过引入抗镰状化人β-球蛋白基因(例如,如pct公布号wo2014043131a1(pct/us2013/059073)中所描述)来治疗镰状细胞病。
上述病变和靶标是说明性的而非限制性的。使用本文提供的教义,本文所述的载体可用于递送大量基因/cdna中的任一种。
干细胞/祖细胞基因疗法。
在各个实施方案中,本文描述的载体特别适用于转导从骨髓、外周血或脐带血获得的人造血祖细胞或造血干细胞(hsc),以及转导cd4+t细胞、外周血b淋巴细胞或t淋巴细胞等。在某些实施方案中,特别优选的靶标是cd34+细胞。在某些实施方案中,优选的靶标是cd34+/cd38-细胞。
当离体转导细胞,例如cd34+细胞、cd34+/cd38-细胞、树突细胞、外周血细胞或肿瘤细胞时,将载体颗粒与所述细胞一起孵育,使用一般大约介于1与50感染复数(moi)之间的剂量,所述剂量也对应于每105个细胞,病毒载体的1×105至50×105个转导单位。这当然包括对应于1、2、3、4、5、6、7、8、9、10、15、20、25、30、35、40、45和50moi的载体的量。通常,载体的量可用hela转导单位(tu)表示。
应注意的是,如pct公布号wo2014043131a1(pct/us2013/059073)中的实施例1所示,仅对于在2×107tu/ml以下的载体浓度发现所实现的基因转移的剂量相关性增加(通过qpcr测量的平均vc/细胞)。更高的载体浓度不会提高转导功效,且实际上通常对转导的程度具有负面影响(数据未示出)。基于这些发现,以2×107tu/ml(moi=40)的标准浓度使用ccl-βas3-fb载体。
在某些实施方案中,基于细胞的疗法包括提供干细胞和/或造血前体,用编码目标转基因(例如,抗镰状化人β-球蛋白)的病毒转导所述细胞,且然后将转化的细胞引入有需要的受试者(例如,具有镰状细胞突变的受试者)。
在某些实施方案中,所述方法包括分离细胞群体(例如来自受试者的干细胞),任选地在组织培养物中扩增细胞,并施用慢病毒载体,所述慢病毒载体在细胞内的存在导致在体外在所述细胞中产生抗镰状化β-球蛋白。然后将细胞返回至受试者,在所述受试者中所述细胞例如可提供产生抗镰状化β球蛋白的红细胞群体,参见例如pct公布号:wo2014043131a1(pct/us2013/059073)中的图16。
在本发明的一些实施方案中,可使用细胞群体,所述细胞群体可以是来自细胞系的细胞或来自除受试者之外的个体的细胞。从受试者分离干细胞、免疫系统细胞等并将所述细胞返回至所述受试者的方法是本领域中熟知的。此类方法被用于例如经历化学疗法的患者中的骨髓移植、外周血干细胞移植等。
在使用干细胞的情况下,将认识到此类细胞可源自多种来源,包括骨髓(bm)、脐带血(cb)cb、动员的外周血干细胞(mpbsc)等。在某些实施方案中,考虑使用诱导型多能干细胞(ipsc)。分离造血干细胞(hsc)、转导此类细胞并将所述细胞引入哺乳动物受试者的方法是本领域的技术人员熟知的。
载体的直接引入。
在某些实施方案中,考虑通过直接引入载体来直接治疗受试者。所述载体组合物可被配制用于通过任何可用的途径递送,所述途径包括但不限于胃肠外(例如,静脉内)、皮内、皮下、口服(例如吸入)、透皮(局部)、透粘膜、经直肠和阴道。常用的递送途径包括吸入、胃肠外和透粘膜。
在各个实施方案中,药物组合物可包含与药学上可接受的载剂组合的载体。如本文所用,表述“药学上可接受的载剂”包括与药物施用相容的溶剂、分散介质、包衣、抗细菌剂和抗真菌剂、等渗剂和吸收延迟剂等。补充活性化合物也可并入所述组合物中。
在一些实施方案中,活性剂(即,本文所述的载体和/或与所述载体一起施用的其他药剂)与将保护化合物免于从身体快速消除的载剂一起制备,所述载剂如控释制剂,包括植入物和微胶囊化的递送系统。可使用可生物降解的、生物相容的聚合物,如乙烯乙酸乙烯酯、聚酐、聚乙醇酸、胶原、聚原酸酯以及聚乳酸。用于制备此类组合物的方法对于本领域的技术人员将是显而易见的。合适的材料也可从alzacorporation和novapharmaceuticals,inc商购。脂质体也可用作药学上可接受的载剂。这些都可根据本领域技术人员已知的方法,如在美国专利号4,522,811中所述的方法来制备。在一些实施方案中,所述组合物被靶向特定细胞类型或被病毒感染的细胞。例如,可使用针对细胞表面标志物(例如在受感染细胞的表面上表达的内源性标志物或病毒抗原)的单克隆抗体来靶向组合物。
有利的是以易于施用和实现剂量均匀性的剂量单位形式配制组合物。如本文所用的剂量单位形式是指适合作为用于待治疗的受试者的单一剂量的物理上离散单位;每个单位包含预定量的经计算结合药用载剂产生所需治疗作用的载体(例如,lv)。
单位剂量不必作为单次注射施用,而是可包括在设定的时间段内连续输注。本文描述的载体的单位剂量可方便地就载体的转导单位(t.u.)进行描述,如通过在细胞系如hela或293上滴定载体所定义。在某些实施方案中,单位剂量可在103、104、105、106、107、108、109、1010、1011、1012、1013t.u.和更高的范围内。
药物组合物可根据需要以不同的时间间隔和在不同的时间段内施用,例如每周一次持续约1至约10周;约2至约8周;约3至约7周;约4周;约5周;约6周等。可能有必要无限期地施用治疗组合物。本领域的技术人员将了解,某些因素可能影响有效治疗受试者所需的剂量和时程,所述因素包括但不限于,疾病或病症的严重程度、先前治疗、受试者的一般健康状况和/或年龄以及存在的其他疾病。如本文所考虑的用载体(例如lv)治疗受试者可包括单一治疗,或者在许多情况下可包括一系列治疗。
用于施用基因疗法载体的说明性剂量和用于确定合适剂量的方法是本领域中已知的。还应理解的是,载体的适当剂量可取决于具体的受体和施用模式。任何特定受试者的合适剂量水平可取决于多种因素,包括所讨论的病变,目标组织,受试者的年龄、体重、总体健康状况、性别和膳食,施用时间,施用途径,排泄速率,其他施用的治疗剂等。
在某些实施方案中,可通过例如静脉内注射、局部施用或立体定向注射来将载体递送至受试者(参见例如,chen等人(1994)proc.natl.acad.sci.usa,91:3054)。在某些实施方案中,载体可口服或经由吸入递送,并且可被包封或以其他方式操作以保护它们免于降解,增强摄取至组织或细胞中等。药物制剂可包含可接受的稀释剂中的载体,或者可包含其中包埋有载体的缓释基质。或者或另外地,当载体可自重组细胞完整地产生时(如本文所述的逆转录病毒载体的情况),药物制剂可包含一种或多种产生载体的细胞。包含本文所述的载体的药物组合物可任选地与用于施用的说明书一起被包括于容器、包装或分配器中。
上述组合物、方法和用途意图是说明性的而非限制性的。使用本文提供的教义,关于所述组合物、方法和用途的其他变化对于本领域的技术人员而言将是可容易获得的。
实施例
提供以下实施例来说明、但非限制所要求保护的本发明。
实施例1
剪接介导的内含子丢失的挽救使含有人泛素c启动子的慢病毒载体中的表达最大化
慢病毒载体几乎普遍使用异源内部启动子来表达转基因。最常用的启动子片段之一是来自人泛素c(ubc)基因的1.2-kb序列,其涵盖ubc的启动子、一些增强子、第一外显子、第一内含子和第二外显子的一小部分。因为在载体产生期间在载体基因组转录后可发生剪接,所以研究了ubc启动子片段内的内含子是否忠实地传递至靶细胞。如本实施例中所描述,基因分析揭示超过80%的原病毒形式缺乏ubc启动子的内含子。在慢病毒包装过程中,人延伸因子1α(eef1a1)启动子片段内含子没有丢失,并且ubc与eef1a1启动子内含子之间的这种差异是由启动子外显子序列赋予的。ubc启动子内含子丢失导致转基因表达的4倍减少。表达盒向相对链的移动阻止内含子丢失并恢复充分表达。这种表达增加主要是由于内含子内的非经典增强子活性,并且推定的内含子增强子序列向多个启动子近端位点的移动实际上抑制了表达。ubc启动子的逆转也阻止了内含子丢失并且在双向慢病毒载体中恢复充分表达。
材料和方法。
质粒构建
这些研究中使用的所有质粒序列都包括在随附的序列表中,所述序列表出于所有目的以引用的方式并入本文。
将人泛素c启动子从fugw扩增(lois等人(2002)science,295:868-872),用t4多核苷酸激酶磷酸化并连接至线性化和平末端化的pcafe(用于表达的盒)以产生pcafe-ubc。将土拨鼠肝炎病毒转录后调控元件序列(本文“pre”,在schambachet等人中称为“lpre”)进行聚合酶链式反应(pcr)扩增并使用in-fusion(clontechlaboratories,mountainview,ca,usa,目录号639645)克隆到用kpni线性化的pcafe-ubc中。将egfp的emerald变体从prset-emgfp(lifetechnologies,carlsbad,ca,usa,目录号v353-20)pcr扩增并使用in-fusion克隆到hpai-线性化的pcafe-ubc-pre中以产生pcafe-ubc-emgfp-pre。以相似的方式产生了pcafe-ubcs-emgfp-pre,其中ubc克隆引物被设计为省去ubc内含子序列。
对于反向取向(ro)质粒pcafe-roubc-emgfp-bghpa和pcafe-roubcs-emgfp-bghpa中的表达盒,从pcdna4/hismaxa(lifetechnologies,目录号v864-20)扩增牛生长激素聚腺苷酸化信号序列,并插入转基因之后。
对于ubc内含子重新定位(i)的构建体pcafe-iubc-emgfp-pre、pcafe-roiubc-emgfp-pre和pcafe-rofiubc-emgfp-pre,从pcafe-ubc-pre扩增ubc内含子序列并使用in-fusion克隆到ecorv-线性化的pcafe-ubcs-emgfp-pre中。
对于ubc增强子缺失(denh)的构建体pcafe-denhubc-emgfp-pre,使用重叠的、位于推定内含子增强子区域侧翼的面向外部的引物扩增pcafe-ubc-emgfp-pre并在dpni处理后用in-fusion再环化。
对于所有pcclc(dull等人(1998)j.virol.,72:8463-8471)lv,将表达盒用ecorv/kpni消化从pcafe质粒中移除并连接到ecorv/kpni-线性化的pcclc中。
通过组装牛生长激素polya(bghpa)和双向mcmv/ubc启动子(kamata等人plosone,5:e11834)以及来自fugw的egfp和wpre的pcr扩增子构建了双向(bd)载体,并且使用in-fusion克隆试剂盒(clontech,mountainview,ca,usa)设计了与pcclc骨架具有重叠同源性的来自efs-single-idlv(jogleka等人(2013)mol.ther.,21:1705-1717)的mcherry。通过限制性消化bd来构建robd载体,以反转反向bghpa与wpre之间的mcherry双向启动子-egfp盒,并用nebquick连接酶试剂盒(newenglandbiolabs,ipswitch,ma,usa)连接。
细胞培养
通过将50-ml热灭活的胎牛血清(geminibio-products,westsacramento,ca,usa,目录号900-208)和5.5-ml100xl-谷氨酰胺:青霉素:链霉素溶液(geminibio-products目录号400-110)添加至500-ml不含l-谷氨酰胺的杜氏改良的伊格尔氏培养基(mediatech,herndon,va,usa,目录号15-013-cv)中来制备d10培养基。通过将相同的两种组分添加至不含l-谷氨酰胺的500-mlrpmi1640培养基(mediatech目录号15-040)中来制备r10培养基。将293t细胞(atcc,manassas,va,usa,目录号crl-1268)维持在d10培养基中,并将k562(atcc目录号ccl-243)细胞维持在r10培养基中。
载体产生
通过用10-μgpcmvδr8.91(zufferey等人(1997)nat.biotechnol.,15:871-875)、10μg的适当pcclc载体质粒和2-μgpcag-vsv-g(hanawa等人(2002)mol.ther.,5:242-251)转染1×107个hek293t细胞来产生lv上清液。通过添加质粒和66-μl1-mg/ml支链pei溶液(sigma-aldrich,st.louis,mo,usa,目录号408727-100ml)且然后涡旋数秒来在1.5-mldpbs中制备转染混合物。在室温下孵育5-10分钟后,将转染混合物逐滴添加至在24小时之前接种于10cm培养皿中的293t细胞。在约16小时后,将培养基更换为补充有50-u/ml青霉素、50-μg/ml链霉素、2-mml-谷氨酰胺和20-mmhepes的ultraculture培养基(lonza,basel,switzerland,目录号12-725f)。在所述培养基更换后第24-48小时收获病毒上清液。
对于roubc载体,包含5-μgpcdna3-novb2以防止由与载体基因组rna反义的转录物的存在而引起的滴度下降(maetzig等人(2010)genether.,17:400-411)。再添加15-μl1-μg/mlpei溶液以补偿增加的质粒dna。
通过在beckmancoultersw-32ti转子中在4℃下在26000rpm下超速离心90分钟来将载体浓缩约150倍,以用于转导人cd34+hspc。
转染
将293t细胞在6孔板(corning,corning,ny,usa,目录号3516)中以8×105个细胞/孔接种于d10培养基中。24小时后,制备1.5μg的质粒用于在1.5-ml微量离心管中的200-μlopti-memi培养基(lifetechnologies,carlsbad,ca,usa,目录号31985-062)中进行转染。添加4.5μl的transit-293转染试剂(mirusbio,madison,wi,usa,目录号mir2700),并将混合物短暂涡旋且在室温下孵育5分钟,然后逐滴添加至细胞。在通过短暂胰蛋白酶消化转染后第48小时收集细胞,并在bdlsrfortessa流式细胞仪上分析绿色荧光蛋白报告蛋白表达。
转导
对于慢病毒表达分析,将k562细胞以50000个细胞/孔接种于24孔板中,并用一系列载体剂量处理以获得具有10%转导或更低的群体,从而确保大部分细胞仅接受单一整合。在流式细胞术分析之前将细胞培养1-2周,以稀释非整合载体并使荧光蛋白水平达到稳定状态。
对于来自动员的外周血的原代人cd34+hspc中的表达分析,将冷冻保存的细胞解冻并在补充有50-ng/ml人flt-3配体、50-ng/ml人干细胞因子和50ng/ml人促血小板生成素(peprotech,rockyhill,nj,usa)的x-vivo15培养基(lonza)中预刺激过夜。然后将病毒载体加入等体积的相同培养基中以达到3x105个转导单位/ml的最终载体浓度,如通过转导k562细胞所测定。这种载体剂量产生约10%的转导。在添加载体24小时后,添加2ml的骨髓分化培养基。所述培养基由补充有20%fbs、0.5%牛血清白蛋白、5-ng/ml人白细胞介素-3、10-ng/ml人白细胞介素-6和25-ng/ml人干细胞因子(peprotech)的imdm组成。
剪接的pcr分析
使用kapahifi热启动聚合酶和引物ubc内含子f(aagtagtcccttctcggcgat,(seqidno:1))、ubc内含子r(ggtcagcttgccgtaggt,(seqidno:2))、eef1a1内含子f(gttctttttcgcaacgggtttg,(seqidno:3))和eef1a1内含子r(tgtggccgtttacgtcgc,(seqidno:4))将来自转导的k562细胞的基因组dna经由pcr进行分析。
使用引物ubcintf(ggcgagtgtgttttgtgaagttt,(seqidno:5))和emgfpr(tacgtcgccgtccagctc,(seqidno:6))以及探针fam-emgfp(fam-caccaccccggtgaacagctcctcg,(seqidno:7))通过分析来自ubc载体转导的k562细胞的基因组dna来进行定量微滴数字pcr(ddpcr)。对于eef1a1载体分析,将ubcintf引物用eef1a1intf(tctcaagcctcagacagtggt,(seqidno:8))取代。
使用ubcsf(gctgtgatcgtcacttgaca,(seqidno:9))代替ubcintf来定量ubc的剪接形式。使用100ng的模板gdna根据制造商的说明书来进行ddpcr。将1单位drai酶(newenglandbiolabs)添加至含有ddpcrsupermixforprobes(bio-rad,hercules,ca,usa)的ddpcrmastermix,并且在37℃下在pcr反应混合物中进行预消化1-2小时,之后进行微滴产生和热循环。
对于分析载体上清液中的载体基因组,使用purelinkrna微型试剂盒液体样品程序(lifetechnologies)从500μl的原始载体上清液中纯化rna。进行反向转染,之后使用iscriptcdna合成试剂盒(bio-rad)进行pcr。
荧光素酶测定
含有由最小tata盒启动子驱动的优化的荧光素酶orf的pgl4.25载体(promega,madison,wi,usa)被用来测定ubc和eef1a1内含子的增强子活性。使用来自cmv启动子的无启动子增强子序列作为阳性对照。经由pcr和gibson组装将所有插入物克隆到用ecorv和kpni线性化的pgl4.25中。在接种于96孔组织培养物处理的平板上的293t细胞中进行荧光素酶测定。将50000个细胞/孔接种在d10培养基中,且在18小时后,在具有100-ng报告质粒和0.3-μltransit-293/孔的opti-mem中制备转染混合物。在用双荧光素酶报告蛋白测定系统(promega)转染后48小时制备样品,并用tecaninfinitem1000pro板读取器(tecan,mannedorf,switzerland)取得发光读数。
结果。
ubc内含子从原病毒形式中缺失,并且表达盒逆转可防止丢失
为了评估ubc内含子1是否在包装期间维持,用ubc启动子的各种修饰产生了用于瞬时转染的pcclclvdna构建体和更简单的pcafe表达质粒构建体(图3)。所有构建体均都含有绿色荧光蛋白的emerald变体(emgfp),其允许经由流式细胞术进行表达分析(tsien(1998)annu.rev.biochem.67:509-544)。ubc构建体含有完整的ubc启动子片段,因为它存在于人基因组中,而较短的ubcs构建体被设计为ubc内含子1完全缺失,如果在包装期间发生典型剪接,则这将是预期的原病毒形式。为了测试表达盒向相对链的移动是否将避免剪接介导的内含子丢失,通过逆转启动子和转基因并在转基因之后插入聚腺苷酸化信号产生了反向取向(ro)构建体roubc和roubcs。重要的是,尽管pcclclv的有效负载穿过rna中间体阶段并且易于经历剪接介导的丢失,但pcafe表达质粒的有效负载没有rna中间体,且因此不会由于剪接而丢失遗传元件。在293t细胞中产生了病毒载体,并用于转导k562细胞以进行原病毒形式的基于pcr的基因分析(图4,图a)。转导后两周gdna的pcr分析揭示,许多cclc-ubcemgfp-pre原病毒形式含有与内含子丢失一致的扩增子,如通过ubcs原病毒形式的分析所指示(图4,图b,泳道5和6)。短产物的sanger测序证实发生了预期典型剪接(数据未示出)。相比之下,roubc原病毒形式不产生截短的pcr产物,从而表明表达盒的逆转完全阻止了内含子丢失(图4,图b,泳道7)。由于含内含子的模板与缺乏内含子的模板之间的预测pcr产物大小的显著差异,可能存在针对缺乏内含子的模板的扩增的显著偏差并且从所述结果过高估计了内含子丢失的量。因此,为了定量内含子丢失的频率,建立了双重数字pcr测定,其中将来自跨越内含子和emgfp转基因的引物和探针组的信号使用引物和探针组标准化至lv包装信号(图5,图a和b)。这一分析表明仅18%的ubc载体形式保留了ubc内含子(图5,图c),而roubc载体形式完全保留了内含子。
为了评估在转导和逆转录期间的事件是否影响含有内含子的原病毒形式的比例,从ubc病毒上清液中收集了rna并定量了含有剪接的ubc内含子的rna基因组的分数。然后,将此分数与含有用相同上清液转导的k562细胞中的剪接的内含子的载体原病毒形式的分数进行比较。这些值非常接近,从而表明内含子已经在载体颗粒中缺失,且因此在包装细胞中被除去(图9)。
内含子的丢失降低了自ubc启动子的表达
为了评估内含子丢失对转基因表达的影响,将含有完整ubc启动子元件或缺失内含子区域的截短的ubc启动子的pcafe表达质粒瞬时转染到293t细胞中,并在转染后第48小时经由流式细胞术进行分析。ubc启动子产生比ubcs启动子显著更高的表达,高约2倍(图6,图a)。在roubc与roubcs构建体之间观察到类似的2倍差异。因为这些质粒被直接转染到细胞中,所以无内含子损失是可能的,并且所测试的ubc启动子质粒都含有内含子。
已经确定了在这些转染实验中内含子的存在赋予更高的表达,其中内含子丢失是不可能的,接下来检查在k562细胞转导2周后来自包装为lv的各种构建体的表达。由于基因分析揭示大多数ubclv形式缺乏内含子,所以推断ubc载体将表达与ubcs载体类似的emgfp水平。事实上,用ubc载体转导的群体中表达emgfp的细胞的荧光与用ubcs载体转导的群体中的荧光几乎相当(图6,图b)。相比之下,roubc载体显示比ubc载体高约2倍的荧光细胞,这与基因分析一致,从而表明roubc载体保留内含子。
还转导了从用粒细胞集落刺激因子处理的健康供体的外周血中富集的人cd34+造血干细胞和祖细胞,以确定在与慢病毒基因疗法相关的原代细胞类型中是否也观察到与ubc载体相比,来自roubc载体的改进的表达。在骨髓分化条件下转导后培养10天后,用roubc载体转导的细胞显示比用ubc转导的细胞高4倍的表达(图10)。基因分析显示,ubc转导的细胞中的内含子丢失与在k562细胞中观察到的内含子丢失相似,并且内含子在roubc转导的细胞中完全维持(图11)。
ubc内含子对表达的积极作用不是由经典的增强子活性引起
除了表达盒的逆转外,还寻求其他方式来保留lv中ubc启动子片段的完整表达。首先研究了与全长ubc启动子片段相比,所报道的内含子增强子序列向紧邻启动子上游的位点的移动是否会产生相等的表达(bianchi等人(2009)gene,448:88-101)。重要的是,这种变体缺乏内含子剪接位点,这应该允许它在lv中传递。然而,所产生的iubc构建表现比ubcs更差(图6,图c)。产生了roiubc和rofiubc并且进行分析以评估增强子序列相对于启动子的取向是否重要,但是这些启动子变体表达并不比iubc好(图6,图c)。最终构建了denhubc,其中使推定的增强子序列缺失,但剪接位点被保留。这种变体表达比ubcs稍微更多的emgfp,这可能是由于来自剪接的核输出改善,但是比ubc显著更少(图6,图c)。这些结果与关于ubc启动子片段内含子的后续研究一致,所述后续研究发现其增强子活性完全依赖于其在内含子内的位置(bianchi等人(2013)plosone,8:e65932)。对这种称为内含子介导的增强的行为知之甚少。
推断,如果ubc内含子序列确实不是经典增强子,则它不应该增加自异源最小启动子的表达。实际上,当内含子序列以正向或反向取向置于最小启动子上游的荧光素酶报道质粒中时,与其中cmv增强子序列置于上游的质粒相比,未观察到相对于背景的荧光素酶表达的增加(图12)。实际上,来自这些质粒的表达显著低于来自仅具有最小启动子的质粒的表达,这与ubc内含子序列被置于转录单元之外时为抑制性的一致。这种抑制作用反映当内含子序列被置于ubcs启动子形式上游时所观察到的表达降低(图6,图c)。有趣的是,处于正向或反向取向的eef1a1内含子1也是如此(图12)。
eef1a1内含子被保持在原病毒形式中并且有助于最大表达
因为观察到自ubc启动子的内含子丢失与关于lv中延伸因子1α(eef1a1)启动子片段的报道形成如此鲜明的对照,所以利用eef1a1启动子片段和emgfp报告基因产生了用于瞬时转染和慢病毒产生的表达载体。来自转导的细胞的gdna的pcr和ddpcr分析显示,几乎所有载体形式均保留启动子内的内含子(图7,图b,泳道5)。凝胶电泳图像的极端对比度调整可揭示在内含子丢失后几乎不可检测量的预期的长度的短产物,但是定量ddpcr分析未检测到缺乏内含子的原病毒形式的这一小群体(图7,图c)。与这些观察结果和先前报道(2)一致,在瞬时转染实验(图7,图d)和转导实验(图7,图e)两者中均观察到含内含子的启动子与缺乏内含子的启动子之间的表达的约2倍差异,从而表明eef1a1启动子元件的内含子确实在几乎所有情况下忠实地完全传递。
内含子传递的差异由启动子外显子序列决定
假设ubc启动子与eef1a1启动子之间的内含子保留的差异是由于内含子内的剪接效率或剪接动力学的序列决定因素所致。为了测试这一点,将内含子从一个启动子交换至另一个启动子,从而产生ubc(eef1a1int)和eef1a1(ubcint)载体。出人意料地,据发现ubc(eef1a1int)lv丢失了eef1a1内含子并且表达与无内含子的ubcs载体类似水平的emgfp,而eef1a1(ubcint)维持了ubc内含子并且表达比无内含子的eef1a1s载体显著更多的emgfp(图13)。这些结果表明两种启动子的不同外显子序列决定内含子是否在慢病毒产生过程中保留。
表达盒修饰使来自ubc双向载体的表达最大化
最终寻求改进来自基于ubc启动子的双向载体的表达,所述双向载体介导lv中的两种转基因的协同表达(kamata等人plosone,5:e11834;amendola等人(2005)nat.biotechnol.,23:108-116)。因为载体设计需要ubc启动子的有义链取向,所以推断大多数原病毒形式将丢失ubc内含子,并且双重的不同ubc和最小巨细胞病毒(cmv)启动子的逆转将导致增加的表达,这是由于内含子包含ubc促进的转基因(图8,图a)。
对稳定转导的293t细胞的基因分析揭示,ubc内含子在75%的时间从bd载体丢失,其中ubc启动子位于载体有义链上,而在robd载体中,几乎所有的原病毒形式都含有ubc内含子(图8,图b)。这导致在稳定转导的细胞中由ubc启动子驱动的egfp表达的增加(图8,图c)。出人意料地,鉴于表明内含子不含传统增强子的表达数据,在robd转导的细胞中保留的ubc内含子中由最小cmv启动子驱动的mcherry表达也增加。
讨论
慢病毒基因转移最近已经进入临床基因疗法试验,取得了多项成功,并且没有临床上显著的不良事件,并且在许多即将进入临床的临床前研究中也显示出前景(cartier等人(2009)science,326(5954):818-823;cavazzana-calvo等人(2010)nature,467:318-322;aiuti等人(2013)science,341(6148):1233151;biffi等人(2013)science,341(6148):1233158;romero等人(2013)j.clin.invest.123(8):3317-3330;carbonaro等人(2014)mol.ther.,22:607-622)。随着疗法被开发用于另外的病症,将产生携带用于转基因调控的各种基因组片段的新载体。过去的启动子/转基因组合需要内含子的存在来获得完整活性和调控,并且很可能一些未来的设计也需要它们。
结果表明内含子在其在载体产生和转导期间丢失的可能性方面不同。虽然人ubc启动子片段在大多数原病毒形式中缺失其内含子,但是人eef1a1启动子片段未类似地受影响。包含ubc内含子需要转基因盒被逆转以避免剪接机构的加工,但是eef1a1内含子几乎在每种原病毒形式中保持,即使它理论上暴露于剪接体(spliceosome)。先前的研究表明杂合cag启动子在整个载体产生和转导中也被保持(ramezani等人(2000)mol.ther.,2:458-469)。eef1a1启动子的无内含子型式已进入腺苷-脱氨酶缺陷型严重联合免疫缺陷病(ada-scid)和x连锁scid(scid-x1)两者的临床试验,并且临床前研究表明,它将驱动足够的转基因表达来获得治疗作用。数据表明,含有内含子1的完整eef1a1启动子产生高约2倍的转基因表达,这是对于未来的载体设计可能是必需或有益的增加。还发现,几乎没有由用该这种载体转导产生的原病毒形式丢失其内含子。总体而言,这些结果说明了逆转录病毒载体的全面基因表征的重要性,因为已知或未知的内含子可能导致携带高度不同的载体形式的转导细胞。在基因疗法领域,在从调控角度来看产物表征是重要的情况下,这种变化不太可能被接受。
据报道,针对剪接供体或受体位点的反义rna可阻止初级转录物的剪接(morcos(2007)biochem.biophys.res.comm.358:521-527)。因此,试图在慢病毒产生期间使用u6驱动的表达针对剪接供体或剪接受体位点的50nt反义序列的质粒来抑制ubc载体基因组的剪接。不幸的是,当单独或组合使用时,这些构建体在转导后未产生来自ubc载体的更高表达(数据未示出)。有可能这种策略可导致在实验中lv中的其他内含子的保留,但是对于ubc内含子是无效的。据发现ubc启动子内含子确实如先前所报道的那样增加表达,但是内含子序列内的增强子样活性依赖于其在内含子内部的位置。这可与未来的载体设计并行,所述载体设计并入具有内源性内含子的转基因获得完整活性,其中内含子序列所包含的调控活性可能类似地不可移动。还批准了进一步的研究以便研究ubc内含子序列当在转录单元内存在时对表达具有积极作用、但是当置于启动子上游时具有负面作用的原因。这可能对用于基因疗法和基因工程的内源性基因调控和转基因调控两者具有重要意义。重要的是,数据表明此类内含子是用于整合载体的相对安全的有效负载,因为它们可能不会以在临床基因疗法试验中引起不良事件和亚临床克隆扩增的方式反式活化附近的启动子(cavazzana-calvo等人(2010)nature,467:318-322;hacein-beyabina等人(2003)science,302:415-419;hacein-bey-abina等人(2008)j.clin.invest.,118:3132-3142;howe等人(2008)j.clin.invest.,118:3143-3150;stein等人(2010)nat.med.,16:198-204)。
ubc内含子和eef1a1内含子1在序列水平上在其对典型剪接供体、受体和分支点位点的附着方面没有显著差异,并且来自交换了内含子的载体的数据表明内含子丢失的序列决定因素不是在内含子本身内,而是在ubc和eef1a1启动子的外显子序列内。如果一般是真实的则这是不幸的,因为在为编码序列的大多数外显子中对载体进行潜在修饰以改变剪接将被显著地限制。从生物学的角度来说,这并不出人意料,因为人基因组中的外显子已知含有外显子剪接增强子以及外显子剪接抑制子/沉默子。这些序列控制人内含子的剪接效率,其中大部分被认为是次优地定义的(zheng(2004)j.biomed.sci.,11:278-294)。
据信这些内含子之间的丢失频率的差异与它们被剪接的速度有关,这可能在很大程度上由外显子序列决定。未来的实验可评估这两种遗传元件的剪接动力学,其速度将被预测与内含子传递反向相关。虽然新的工作已经检查了转录剪接和从染色质释放的动力学,但是对于不同转录物观察到的速率范围的序列决定因素尚不清楚(pandya-jones等人(2013)rna,19:811-827)。对剪接动力学的决定因素的更好理解可指导ubc启动子片段的修饰以充分降低剪接速度来使含有内含子的基因组rna进入载体颗粒中,同时在转基因表达期间维持有效剪接。
实施例2
人造血干细胞/祖细胞的富集促进用于干细胞基因疗法的转导
用于镰状细胞病的自体造血干细胞(hsc)基因疗法具有治疗这种疾病的潜力,而没有与同种异体移植相关的主要免疫学并发症。然而,使用富含cd34的细胞群体的由b-球蛋白慢病毒载体进行的转导效率不是最理想的,并且临床试验可能需要较大载体生产批量。转导更加富含hsc的细胞群体可大大降低载体需要,并且可能提高转导效率。通过荧光活化的细胞分选从健康的脐带血cd34+细胞中分离组成所有cd34+细胞的约1%-3%的cd34+/cd38-细胞,并用表达β-球蛋白的抗镰状化形式的慢病毒载体(ccl-βas3-fb)转导。与cd34+细胞相比,分离的cd34+/cd38-细胞能够在长期培养(ltc)的延长时间段内产生子代,并且与含有相等数量的cd34+/cd38-细胞的本体cd34+制剂相比需要少达40倍的载体来转导。通过定量pcr在第14天测量,分离的cd34+/cd38-细胞的转导与cd34+细胞相当,其中载体需要减少,并且对于从cd34+/38-细胞起始的ltc,随时间推移的平均载体拷贝/细胞保持较高。在体外红系细胞分化后,来源于cd34+/cd38-细胞或未分化的cd34+细胞的培养物中的hbbas3mrna表达相似。体内研究显示当与100倍以上的cd34+/cd38+细胞竞争移植时,转导的cd34+/cd38-细胞的等效植入。这项工作提供了分离人cd34+/cd38-细胞以使用显著更少的载体并潜在改进hsc基因疗法的转导的有益作用的证据。
引言
基于造血干细胞的疗法可潜在治疗许多遗传性和获得性血细胞疾病,并且近年来已经取得了令人兴奋的临床进展(kohn等人(2013)biol.bloodmarrowtransplant.19(增刊1):s64-s69)。镰状细胞病(scd)是与严重急性疾病和进行性器官损伤相关的多系统疾病,其导致显著发病率和早期死亡率(platt等人(1994)n.engl.j.med.330:1639-1644)。它是全世界最常见的遗传病症之一,在美国影响大约90,000人。当前治疗主要包括贫血和疼痛的对症疗法。羟基脲(hu)是在低氧张力条件下诱导胎儿血红蛋白(hbf)产生以抑制镰状血红蛋白(hbs)的聚合的另一种治疗选择;然而,由于各种原因,hu未被广泛使用(green和barral(2014)pediatr.res.75:196-204;brandow等人(2013)am.j.hematol.85:611-613)。scd的唯一潜在治疗是异基因造血干细胞移植(hsct)。这通常需要良好匹配的供体,并且可伴随着对长期免疫抑制的需要以及移植排斥或移植物抗宿主疾病的可能性,但是最近关于匹配的同胞干细胞的成年受体中强度有效降低的病状的报道带来希望(hsieh等人(2014)jama312:48-56)。
使用自体造血干细胞(hsc)的基因疗法是用于scd的有希望的治疗,可能没有在异体hsct情况下观察到的主要免疫并发症(chandrakasan等人(2014)hematol.oncol.clin.northam.28:199-216)。已显示当添加至小鼠和人造血干细胞/祖细胞(hspc)中时抗镰状化βas3-球蛋白基因具有与hbf相似的抑制红细胞(rbc)镰状化并防止scd表现的活性(levasseur等人(2003)blood102:4312-4319;levasseur等人(2004)j.biol.chem.279:27518-27524;romero等人(2013)j.clin.invest.123:3317-3330)。然而,携带复杂且相对较大的人β-球蛋白基因组表达盒的慢病毒载体具有低滴度;人cd34+hspc的转导仅适度有效且需要使用相对大量的载体,同时产生相对较低的基因转移(例如,每个细胞的平均载体拷贝数为0.5-1.0)。未浓缩的生产批量产生≤106个转导单位(tu)/ml的滴度,从而需要产生大量的载体来进行临床规模的转导。理想地,鉴别使用较少病毒载体的方法将避免高载体产生成本并允许更多患者的治疗(logan等人(2004)hum.genether.15:976-988)。
cd38是在各种成熟的造血细胞上表达的ii型膜表面糖蛋白。在早期hspc群体上cd38表达较低或不存在(hao等人(1995)blood,86:3745-3753;hao等人(1996)blood,88:3306-3313;albeniz等人(2012)oncol.lett.3:55-60),并且原始多能人hsc的大多数定义包含在cd34+/cd38-部分内(notta等人(2011)science,333:218-221)。cd34+/cd38-细胞仅组成cd34+细胞的约1%-3%,且因此对hspc的富集比未分级分离的cd34+群体高50-100倍。cd34+/cd38-细胞具有超过未分级分离的cd34+细胞的长期增殖和血细胞产生的能力(hao等人(1995)blood,86:3745-3753;hao等人(1996)blood,88:3306-3313)。先前公开的研究已经证明了使用相对高的载体浓度用ccl-βas3-fblv载体(romero等人(2013)j.clin.invest.123:3317-3330)以中等效率转导原代人bmcd34+细胞的能力。据推测,通过分离cd34+/cd38-细胞进一步纯化超出标准cd34+细胞富集级分的hspc将减少待离体处理的靶细胞的绝对数量,并且每个治疗的受试者将需要显著更少的载体。
在此,显示使用荧光活化的细胞分选(facs)分离的人脐带血(cb)cd34+/cd38-细胞可用至多40倍的病毒载体转导,并且仍然实现与在未分级分离的cd34+细胞群体中观察到的相当或更高的载体拷贝数目(vcn)。这些结果证明使用富含cd34+/cd38-的hspc改进scd的基因疗法中hsc的转导以获得增加的功效。
材料和方法。
载体
已经描述了ccl-βas3-fb和ccl-mnd-gfp的构建(romero等人(2013)j.clin.invest.123:3317-3330)。ccl-ubiq-mcitrine-pre-fb-2xuse、ccl-ubiqmstrawberry-pre-fb-2xuse和ccl-ubiq-mcerulean-pre-fb-2xuse是使用ccl载体骨架(zufferey等人(1998)j.virol.72:9873-9880)、人泛素启动子(lois等人(2002)science,295:868-872)、购自addgene(cambridge,ma)的荧光基因、优化的转录后调控元件(pre;(zanta-boussif等人(2009)genether.16:605-619;schambach等人(2006)genether.13:641-645))和sv40聚腺苷酸化增强子序列use的两个串联拷贝(schambach等人(2007)mol.ther.15:1167-1173)构建的。将慢病毒载体用vsv-g假型包装,并且如所描述(cooper等人(2011)j.virol.meth.177:1-9)进行浓缩和滴定。还使用rd114/tr质粒用rd-114假型包装了ccl-βas3-fb(sandrin等人(2002)blood100:823-832;bell等人(2010)exp.biol.med.234:1269-1276;rasko等人(1999)proc.natl.acad.sci.usa,96:2129-2134)并如所描述进行滴定(cooper等人(2011)j.virol.meth.177:1-9)。与浓缩的rd114/tr假型制剂的2.7x107tu/ml相比,vsv-g假型ccl-βas3-fb的不同制剂在300-1,000x浓缩后具有的滴度为6x108-6x109tu/ml。
样品收集
在ucla医学中心(洛杉矶)进行的阴道分娩和剖腹产后,在夹紧且切割脐带后通过将血液从胎盘引流至含抗凝柠檬酸盐-磷酸盐-右旋糖的无菌采集管中来获得脐带cb。所有cb试样都是根据加利福尼亚大学批准的指导方针获得的,并一直被视为无名医疗废物而免于irb审查。细胞在收集48小时内进行处理。使用ficollhypaque(stemcelltechnologies,vancouver,bc,canada)密度离心从cb分离单核细胞(mnc)。然后使用免疫磁性柱分离来通过在4℃下将mnc与抗cd34微珠(miltenyibiotec,inc.,bergischgladbach,germany)一起孵育30分钟来富集cd34+细胞。然后使所述细胞通过免疫磁性柱并收集cd34+细胞。将cd34+细胞置于具有冷冻培养基(10%二甲亚砜[sigmaaldrich,st.louis,mo],90%fbs)的冷冻小瓶中并在液氮中冷冻保存直至需要。
荧光抗体标记和cd34+/cd38-细胞分选
将cd34+细胞解冻,洗涤并重新悬浮于75ml的磷酸盐缓冲盐水(pbs)中以与荧光标记的抗体一起孵育。添加未稀释的藻红蛋白(pe)缀合的抗cd38(20ml)和未稀释的别藻蓝蛋白(apc)缀合的抗cd34(5ml)(所有抗体来自bdsciences,sanjose,ca),并将所述细胞在4℃下在黑暗中孵育30分钟。在孵育后,将细胞在pbs中洗涤一次。在facsariaii(bdbiosciences)上进行facs。
活mnc群体由前向散射和4',6-二脒基-2-苯基吲哚(lifetechnologies,grandisland,ny)染色进行设门。使用设门区域来限定cd34+细胞群(图14,图a)。p3用于限定f1cd34+/38-细胞群体,其是对pe阴性的2.5%的apc阳性细胞。p5用于限定对apc阳性且对pe阳性的cd34+/cd38+细胞。这些设门策略用于所有分选实验。
慢病毒载体转导
在细胞分选后,将cd34+和cd34+/cd38-细胞以6.3x104-7.5x105个细胞/毫升的细胞密度置于用retronectin(20mg/mlretronectin,takarashuzo,co.,japan)涂覆的非组织培养物处理的平板的各孔中。在转导培养基(含有1xl-谷氨酰胺/青霉素/链霉素(l-glut/pen/strep)(geminibio-products,westsacramento,ca)、50ng/ml人干细胞因子(hscf)(stemgent,cambridge,ma)、20ng/ml人白细胞介素-3(hil-3)(r&dsystems,minneapolis,mn)、50ng/ml人促血小板生成素(r&dsystems)和50ng/ml人flt-3配体(flt-3)(peprotech,rockyhill,nj)的(无血清x-vivo15培养基(lonza,basel,switzerland))在37℃、5%co2下进行预刺激持续18-24小时。在预刺激后,以指定的载体浓度(通常2x107tu/ml,除非另有说明)将所需病毒载体(ccl-βas3-fb、cclmnd-gfp、mstrawberry、mcerulean、mcitrine或βas3-fb-rd114)添加至各孔且再次在37℃、5%co2下孵育24小时。然后洗涤细胞并将其转移至组织培养物处理的平板以用于在具有5ng/mlil-3、10ng/mlil-6和25ng/mlhscf的基础骨髓培养基(伊斯科夫氏改良的杜氏培养基(imdm)(lifetechnologies,grandisland,ny)、1xl-glut/pen/strep、20%fbs、0.52%牛血清白蛋白(bsa))中在37℃、5%co2下的骨髓分化。根据需要在14天时间段内添加新鲜培养基。在培养14天后,通过qpcr或数字微滴pcr(ddpcr)(hindson等人(2011)anal.chem.15:8604-8610)测定vcn,并使用流式细胞术分析荧光报告基因表达。
长期基质培养物和甲基纤维素培养物
在计划的cd34+细胞分选之前两天,将ms5鼠基质细胞(suzuki等人(1991)leukemia,6:452-458)解冻,以10,000cgy照射,且然后在96孔板中接种(3x104个细胞/孔)于基质培养基(imdm[lifetechnologiesgrandisland,ny],fbs10%,2-巯基乙醇)中以形成预先建立的长期培养物(ltc)的基质层。将分选的cd34+和cd34+/cd38-细胞在ltc培养基(imdm、30%fbs、10%bsa、2-巯基乙醇、106mol/l氢化可的松、1xl-glut/pen/strep连同10ng/ml白细胞介素-3[il-3]、50u/mlil-6和50ng/ml人干细胞因子(hscf))中的经照射的基质上共培养(hao等人(1996)blood,88:3306-3313;breems等人(1998)blood,91:111-117;koller等人(1995)blood,86:1784-1793;bennaceur-giscelli等人(2001)blood,97:435-441)。以2-4周时间间隔,从培养物中取出未附着细胞的样品。如下文所述,用血细胞计数器(thermofisherscientific,pittsburgh,pa)通过活细胞计数测定它们的数量,并使用purelink基因组dna微型试剂盒(invitrogen,carlsbad,ca)提取基因组dna以用于qpcr。
分化和mrna表达分析
体外红系细胞分化测定是基于从如由romero等人(2013)j.clin.invest.123:3317-3330修订的douay和giarratana(2009)meth.mol.biol.482:127-140改编的方案。在预刺激并转导facs分离的cd34+/cd38-细胞和未分级分离的cd34+细胞后,将所述细胞转移至红系细胞培养物。使用lv原病毒中的hiv-1包装信号序列psi的qpcr来分析vcn,并标准化至人细胞常染色体基因多配体聚糖4(sdc4)以计算vcn,如cooper等人(2011)j.virol.meth.177:1-9所描述。如之前由romero等人(2013)j.clin.invest.123:3317-3330所描述测定hbβas3mrna表达。
在免疫缺陷小鼠中移植转导的人cbcd34+/cd38-细胞
将来自健康供体cb的未分级分离的cd34+、cd34+/cd38+和cd34+/cd38-细胞分别用全部在23×107tu/ml(moi5-140)下的携带不同荧光标记基因的慢病毒载体转导。在250cgy全身照射后,通过尾静脉注射将mock转导的(5x105)细胞和对照未分级分离的转导的cd34+(5x105)细胞单独移植到6-10周龄免疫缺陷型nod.cg-prkdscidil2rgtm1wjil/szj(nsg)小鼠(jacksonlaboratory,sacramento,ca)。在照射后将cd34+/cd38-和cd34+/cd38+细胞以1:99比率混合,以使得照射后将2x103个cd34+/cd38-细胞和2x105个cd34+/cd38+细胞通过尾静脉注射共同移植到6-10周龄nsg小鼠中。
在8-12周后,使小鼠安乐死并使用apc缀合的抗人cd45对比horizonv450缀合的抗鼠cd45(bdbiosciences)通过流式细胞术分析bm的人细胞植入。在抗体孵育后,使用bdfacs-溶解溶液(bdbiosciences)溶解解rbc。移植人细胞的百分比被定义为%hucd45+/%hucd45++%mucd45+细胞)。在hucd45+细胞中,使用流式细胞术分析mcitrine、mstrawberry和mcerulean的表达。
使用ddpcr在具有人细胞的阳性植入物的鼠bm样品中测量平均vc/人细胞(表4)。制备反应混合物,所述反应混合物由含有1xddpcrmastermix(bio-rad,hercules,ca)、对hiv-1psi区域具有特异性(以检测所有载体)或对每种荧光报告基因具有特异性的引物和探针(对于引物和探针,分别400nm和100nm)、drai(40u;newenglandbiolabs,ipswich,ma)和1.1ml(对于cfu4ml)基因组dna样品的22ml体积组成。如hindson等人(2011)anal.chem.15:8604-8610中所描述进行微滴生成。热循环条件包括95℃10分钟,94℃30秒和60℃1分钟(55个循环),98℃10分钟(1个循环)和12℃保持(表3)。使用针对常染色体人基因sdc4基因的引物,针对从nsg小鼠收集的骨髓中存在的人细胞的百分比而标准化所述基因的特异性扩增部分,以调节样品中鼠细胞的存在。参与实验的所有动物都根据由ucla动物研究委员会实验室医学部(uclaanimalresearchcommitteeunderthedivisionoflaboratorymedicine)批准的方案进行护理和处理。
表2移植到nod.cg-prkdscidil2rgtm1wjil/szj小鼠中的细胞的体外载体拷贝数目。
表3.pcr引物。
低密度脂蛋白受体表达分析
收集细胞并如先前在荧光抗体染色和细胞分选部分中所描述进行分选。将cd34+和cd34+/cd38-细胞在37℃、5%co2下接种到转导培养基中用retronectin(20mg/mlretronectin,takarashuzo,co.,japan)涂覆的非组织培养物处理的平板的各个孔中。使用流式细胞术,在培养24小时和48小时时,收获细胞以进行低密度脂蛋白(ldl)受体表达的分析,与在培养之前(0小时)的细胞相比较。将细胞洗涤并重新悬浮于90ml的pbs中以与荧光标记的抗体、10ml未稀释的apc缀合的抗人ldl受体(r&dsystems,minneapolis,mn)一起孵育。将细胞在4℃下在黑暗中孵育30分钟。在孵育后,将细胞在pbs中洗涤一次,并用lsrfortessa分析进行分析。apc阳性细胞被认为是对ldl受体表达呈阳性的。
统计分析
在图中给出连续结果变量,如实验条件的平均值和se。双向比较通过单因素或双引物anova的框架内非配对t检验进行。当不符合常态性假设时,通过wilcoxon秩和检验进行两组比较。混合线性模型被用来随时间推移比较两组。0.05的p值被用作显著性阈值。
结果。
使用facs分离cbcd34+/cd38-细胞
使用免疫磁性柱使健康供体cb富集cd34+细胞。通过流式细胞术来分选未分级分离的cd34+细胞的部分以分离cd34+/cd38-细胞,所述cd34+/cd38-细胞在操作上定义为对于cd38表达为最低2.5%的细胞(图14,图a)。以从cb单位分离的2.2-6.8x106个cd34+细胞开始,分离了6x103-6x104个(平均值=2.5x104)cd34+/cd38-细胞,从而代表理论产率的36%-99%的范围(n=11)。
当进入长期培养时,未分级分离的cd34+细胞在第一个月扩增约10倍,且然后数量减少(图1b)。用cd34+/cd38-细胞起始的ltc扩增更大程度(约100倍)并且保持稳定细胞数目超过3个月(图14,图b),从而证明与本体cd34+细胞相比,更原始的cd34+/cd38-群体的更大生成能力。
cbcd34+细胞对比cd34+/cd38-细胞的转导的评估
比较用ccl-βas3-fb慢病毒载体对来自健康供体(n511)的cb的cd34+细胞和cd34+/cd38-细胞的转导。细胞密度和载体浓度以及因此感染复数(moi)对于实验内的两种细胞类型保持恒定,使用在相同体积中的相对数量的cd34+细胞和cd34+/cd38-细胞或在转导不同的细胞数目时调节培养物的总体积。将转导的细胞在短期体外骨髓分化条件下培养2周,在甲基纤维素集落形成单位(cfu)测定中生长(14天)或在长期骨髓培养物中生长(90天)以比较集落形成能力和vcn。
在第14天通过定量(qpcr)针对载体的hiv-1psi区域对从细胞分离的基因组dna进行分析以确定平均载体拷贝数目/细胞(vcn)。在每个样品中,cd34+/cd38-细胞的转导等于或大于cd34+细胞的转导(表2)。与来自转导的cd34+培养物的1.25±0.28相比,由转导的cd34+/cd38-细胞产生的细胞具有显著更高的vcn(2.43±0.41)(n=11,p=.02)(图15,图a)。
由cd34+细胞和cd34+/cd38-细胞形成的集落的类型没有差别(图15,图b)。由接种于甲基纤维素中的25.7%的非转导的cd34+(nt-cd34+)细胞、24.3%的转导的cd34+细胞和22.3%的转导的cd34+/38-细胞形成集落(图15,图c)。用于检测和定量ccl-βas3-fb载体序列的单个cfu的qpcr证明来自cd34+/cd38-细胞的转导的集落形成祖细胞的百分比(73.8%,平均vcn=2.12)高于来自cbcd34+细胞(56.2%,平均vcn=1.75)(n=80个集落,各自)(图15,图d)(p=.52)。与来自未分级分离的cd34+细胞形成的cfu(36.2%)相比,由cd34+/38-细胞形成的cfu显示1-2vc/细胞(47.5%)的更高集落百分比(图15,图d)。
进行载体剂量-反应实验以检查ccl-βas3-fb载体转导人cbcd34+细胞和cd34+/cd38-细胞的相对能力,在转导期间使用从2x106至2x107tu/ml的一系列载体浓度。在两种细胞群体中在增加的载体浓度情况下均观察到在第14天基因转移的剂量相关性增加(通过qpcr测量的vcn)(图15,图e)。然而,在所测试的每一载体浓度下,所得到的vcn对于cd34+/cd38-细胞(在6.6x106tu/ml下p=.05,在2x107tu/ml下p=.002)比对于cd34+细胞高;因此可使用相当更低浓度的病毒载体(2x106tu/ml)来转导cd34+/cd38-细胞,并且仍然与更高载体浓度(2x107tu/ml)下用cd34+细胞实现的转导水平相匹配(图15,图e)。
使转导的cbcd34+细胞和cd34+/cd38-细胞(n=3)在ms5基质细胞上在ltc中生长90天,并且在若干时间点分析细胞样品的vcn(图15,图f)。在每个时间点,与来自未分级分离的cd34+群体的培养物(0.3-0.6)相比,来自cd34+/cd38-细胞的培养物中存在更高的vcn(1.6-2.3)(p=0.0004),具有统计学显著的时间趋势差异(p=.03)。
与从cd34+细胞起始的培养物相比,从cd34+/cd38-细胞起始的ltc具有越来越高的集落形成细胞频率。在ltc的第30天,0.05%的来自ntcd34+细胞的培养物的细胞、0.04%的转导的cd34+以及接种于甲基纤维素中的0.13%的转导的cd34+/38-细胞(p<0.0001)产生集落(图20)。在ltc的第60天,由以下产生了集落:源自ntcd34+细胞的0.0017%,来自转导的cd34+的0.0025%,以及来自接种于甲基纤维素中的转导的cd34+/cd38-细胞的0.0067%(图21)。
从cd34+/cd38-细胞起始的培养物中也呈现比从cd34+细胞起始的那些更高百分比的转导的集落形成祖细胞。当通过ddpcr在ltc的第30天进行分析时,与源自未分级分离的cd34+的培养物的单个cfu的77.3%(平均vcn=2.2)相比,由cd34+/cd38-细胞形成的83.7%(平均vcn52.3)对于ccl-βas3-fb载体呈阳性(图20)。在60天时,未分级分离的cd34+细胞仅产生三个集落,其中一个vcn为0.5,而另外两个vcn为0.0。由cd34+/cd38-细胞形成22个cfu并且16个(88.8%)对ccl-βas3-fb载体呈阳性(平均vcn=1.52)(图21)。
为了确定cd34+/cd38-细胞的更高转导是否将在除ccl-βas3-fb载体以外的载体情况下发生,在剂量反应实验中评估了高滴度的表达绿色荧光蛋白(gfp)的lv载体(ccl-mnd-gfp)对cd34+和cd34+/cd38-细胞的转导效率。在2x106、6.6x106和2x107tu/ml(分别moi=4、40、400)的浓度下用ccl-mnd-gfplv载体以相同的细胞密度转导两个细胞群体。在两种细胞群体中在增加的载体浓度情况下均观察到在第14天基因转移的剂量相关性增加(通过ddpcr测量的vcn)(图16,图a)。在6.6x106和2x107tu/ml的载体浓度下,所得到的vcn对于cd34+/cd38-细胞(cd34+平均vcn=2.25±0.15对比在6.6x106tu/ml下1.36±0.05和4.32±0.6对比3.37±0.07,p=.02在2x107tu/ml)比对于cd34+细胞高。在第14天,使用流式细胞术测定表达gfp的细胞的百分比(图16,图b)并且与cd34+/cd38-细胞中的45%-97%相比(图16,图c),cd34+细胞中在45%至81%的范围内,其没有显著差异,p=0.29。
cbcd34+/cd38-细胞的体外红系细胞分化
为了比较转导cd34+细胞和cd34+/cd38-细胞后ccl-βas3-fb载体的βas3-球蛋白表达,将转导的细胞置于体外红系细胞分化模型(douay和giarratana(2009)meth.mol.biol.482:127-140)中以产生支持β-球蛋白基因盒表达的成熟rbc。使用定量逆转录酶聚合酶链式反应(qrt-pcr)来测量载体表达以特异性地定量来自载体的hbβas3转录物和总b-球蛋白样转录物(内源性hbb和hbβas3)。用ccl-βas3-fblv载体转导来自健康cb供体的cd34+/cd38-细胞和cd34+细胞,并将对照样品进行mock转导。在24小时后,所述细胞分化成红系细胞持续27天。通过用针对红系细胞膜糖蛋白gpa的抗体和dna标记荧光染料draq5进行双重染色,在分化结束时(第27天)鉴定无核rbc。无核rbc被定义为gpa+/draq5-(图22)。最终细胞数目、分化标志物和无核百分比在两个细胞群体之间相似,来自未分级分离的cd34+细胞的52.6%±1.06%的无核红细胞和来自cd34+/cd38-细胞的52.7%±1.08%(p=.87)。
与cd34+细胞的子代相比,cd34+/cd38-细胞的红系细胞子代中存在更高的vcn。在培养2周时,来自cd34+/cd38-细胞的培养物的平均vcn为3.08±0.71,而来自cd34+细胞的平均vcn为1.84±0.44(n=3,p=.26)(图17,图a)。
将用ccl-βas3-fblv转导并在在红系细胞分化培养物测定的第14天收集的细胞通过qrt-pcr针对其hbβas3mrna表达进行评估,并与来自内源性成人hbbmrna的表达进行比较。在所有培养物中表达水平是相似的;hbβas3mrna水平占来自cbcd34+/38-细胞的培养物的红系细胞中的总hbb样mrna的55.2%±18.1%,相比之下占来自cbcd34+细胞的培养物45.4%±16.7%(p=0.59)(图17,图b),这与在健康和scd骨髓cd34+细胞中使用ccl-βas3-fblv的先前研究中报道的量类似(romero等人(2013)j.clin.invest.123:3317-3330)。
在预刺激和转导后ldl受体表达的评估
评价了vsv-g-假型载体(finkelstein等人(2013)proc.natl.acad.sci.usa,110:7306-7311)的最近鉴定的细胞受体人ldl受体中的差异是否是与cd34+细胞相比更有效地转导cd34+/cd38-细胞的原因。使用流式细胞术来分析用apc抗人ldl受体抗体染色的新鲜cbcd34+和cd34+/cd38-细胞中的ldl受体表达,且然后在具有多种细胞因子的转导培养基中培养24和48小时后再次(分别与预刺激和转导完成相关的时间点)。
与新鲜cd34+/cd38-细胞的4.8%相比,新鲜cd34+细胞(7.9%)表达ldl受体(图18,图a、d)。在24小时,约91%的cd34+细胞表达ldl受体,相较于cd34+/cd38-细胞的74.6%(图18,图b,18,图e)。在48小时,ldl受体在99%的cd34+细胞和90%的cd34+/cd38-细胞上表达(图18,图c、f)。ldl受体的几何平均荧光强度在cd34+细胞和cd34+/cd38-细胞上类似(分别1.6x103-3x103和1.5x103-3.5x103)。新鲜cd34+细胞和cd34+/cd38-细胞具有类似的表达ldl受体的低百分比,并且它在用于预刺激和转导的条件下在这些细胞上由培养同等地诱导。因此,ldl受体表达的差异不是cd34+/cd38-细胞的更好转导的基础。
rd114逆转录病毒包膜假型lv
为了确定cd34+/cd38-细胞的改进的转导是否与vsv-g包膜特异性有关,使用rd114逆转录病毒包膜,产生了一批具有交替假型的ccl-βas3-fblv(sandrin等人(2002)blood100:823-832;bell等人(2010)exp.biol.med.234:1269-1276)。与在相同条件下转导且分析的cd34+细胞(0.006±0.05,n=3)相比,用rd114-假型ccl-βas3-fblv载体转导且然后培养14天的cd34+/cd38-细胞具有更高的平均vcn(0.86±0.46)(图18,图g)。因此,cd34+/cd38-细胞的更高转导不是对vsvg假型特异性的。
体内cd34+/cd38-细胞的植入潜力的评估
先前研究将通过facs分离的cd34+/cd38-细胞与未分级分离的cd34+细胞的转导进行比较。对于后续研究,将cd34+/cd38-细胞和cd34+/cd38+细胞的转导和植入潜力进行了比较,所述细胞两者均通过facs分离自未分级分离的cd34+的同一群体。将每种群体用携带不同荧光报告基因的慢病毒载体(cclc-ubc-mcitrine-pre-fb-2xuselv、cclc-ubc-mcerulean-pre-fb-2xuselv和cclc-ubc-mstrawberry-pre-fb-2xuselv载体)进行转导。为了确保结果不受载体影响,用于转导每种细胞级分的载体针对每个研究交替(n=3)。将转导的cd34+细胞、cd34+/cd38+细胞和cd34+/cd38-细胞以其适当的生理比例(99%cd34+/cd38+细胞+1%cd34+/cd38-细胞;或100%cd34+细胞)异种移植到nsg小鼠中。转导条件与用于体外分析相同,并且将所述细胞在24小时转导后立即移植。每只小鼠接受由(a)ntcd34+细胞(mock)、(b)截短的cd34+细胞(对照)或(c)转导的cd34+/cd38+与转导的cd34+/cd38-细胞的组合(测试)组成的5x105个细胞的总细胞剂量,所述细胞各自与作为“填充剂(filler)”的照射的(10,000cgy)cd34-细胞混合(表5)。将小鼠在移植后80-90天安乐死并收获其骨髓以通过流式细胞术分析人细胞的植入物并测量植入细胞的vcn。植入百分比被定义为总cd45+群体(鼠cd45+加人cd45+)细胞中人cd45+细胞的百分比。然后基于所使用的荧光标记和通过ddpcr,通过流式细胞术进一步分析来自人植入小鼠的bm的在人植入细胞中存在的不同的转导的细胞级分的百分比。
进行了三组体内小鼠移植,各自由6只小鼠组成,总计移植了18只小鼠(5只mock[ntcd34+细胞]、4只对照[转导的cd34+细胞]、9只测试小鼠[转导的cd34+/38+与转导的cd34+/38-细胞的混合物])(表5)。使一部分转导的细胞体外生长2周并针对vcn进行测定(表4)。ddpcr引物和探针被设计用于特异性地检测每种荧光标记基因(表3)。在体外培养2周后使用hiv-1psi区引物对转导的细胞的样品进行分析以测量所有载体显示cd34+/38-细胞一致地具有比未分级分离的cd34+细胞(vcn=2.19±0.47)或cd34+/cd38+细胞(vcn=1.66±0.36)更高的vcn(6.89±0.75),不管用于转导的载体是什么(mcitrine、mstrawberry、mcerulean)(表4)。在总计18只小鼠中,14只在bm收获时具有人cd45+细胞的成功植入(图23)。在植入的小鼠中,小鼠#1、6和7接受仅mock转导的人cd34+细胞。对照小鼠#8、9、12接受通过单一载体转导的cd34+的细胞。小鼠#2、3、4、5、10、11、13和14接受用不同载体转导的cd34+/cd38-细胞与cd34+/cd38+细胞的测试混合物(1:99的细胞比率)。总之,与未分级分离的cd34+细胞相比,具有用cd34+/cd38-细胞更好植入的趋势(p=.06)。
表4.移植到nod.cg-prkdscidii2rgtm1wjil/szj小鼠中的细胞的体外载体拷贝数目。
表5.异种移植研究的实验设计--细胞剂量和荧光标记
在八只植入测试小鼠中,在六只小鼠(#2、3、4、5、11和14)中存在载体标记的cd34+/cd38-细胞的更高植入,一只小鼠中(#10)中cd34+/cd38+细胞和cd34+/cd38-细胞的等效植入,以及一只小鼠(#13)中更高的cd34+/cd38+植入(图19,图a,图23)。总体上,通过移植的本体cd34+细胞或分级分离的cd34+/cd38-细胞和cd34+/cd38-细胞的基因标记水平没有差异(表6)且因此转导的cd34+/cd38-细胞对植入的效力比未分级分离的cd34+细胞或cd34+/cd38+细胞高大约100倍。
表6.来自nsg小鼠的骨髓的针对用于标记单独移植细胞群体的每种特异性荧光标记基因和针对总载体原病毒(psi)的数字微滴pcr(ddpcr),标准化至每种骨髓样品中的人基因组。
用单体载体转导的本体cd34+细胞移植的两只小鼠具有的vcn为12和6,使用hiv-1psi区引物或用荧光标记特异性引物测得类似的值。用cd34+/cd38+细胞与cd34+/cd38-细胞的混合物移植的小鼠显示类似水平的用两种载体的基因标记(分别3.60±0.26和2.17±0.15),并且在每只小鼠中,两种单独载体的vcn的和与使用psi区引物测得的总vcn类似(图19,图b)。
讨论。
干细胞基因疗法朝向用于多种疾病(包括scd)的临床进展。因为它对scd是有效的,所以转导必须用足够数目的转导以表达足够的βas3-球蛋白的hsc来有效改变疾病的病理生理学。临床规模的hsc转导可以是因被递送和插入的大型复杂的基因盒如βas3-球蛋白基因而变得更困难的具有挑战性的过程。虽然已经展示了用ccl-βas3-fblv载体转导人cd34+hspc(romero等人(2013)j.clin.invest.123:3317-3330),但是由于病毒载体的次最优未浓缩的滴度,需要较高体积的病毒载体来达到基因转移至植入hsc以校正scd的rbc疾病表现的水平。由于这些原因,可使用较少病毒载体转导以实现相当的基因转导效率的具有有效植入能力的替代细胞群体是吸引人的。迄今为止,若干研究已经表明,cd34+/cd38-细胞可用lv载体进行转导,因为与c-逆转录病毒不同,这些载体能够转导未活跃分裂的细胞(case等人(1999)proc.natl.acad.sci.usa,96:2988-2993;geronimi等人(2003)stemcells,21:472-480;guenechea等人(2000)mol.ther.1:566-573)。然而,尚未探索cd34+/cd38-细胞被转导和植入用于基因疗法的能力,这是由于临床等级试剂的缺乏和大规模gmp细胞分选的挑战。
进行了使用人cbcd34+/cd38-细胞来评估这些细胞用于scd的基因疗法的潜在适合性、同时使用较少病毒载体用于转导的研究。研究已经表明,分离自cb的cd34+/cd38-细胞易于用慢病毒载体转导,从而需要显著更低量的病毒载体来实现与cd34+细胞相比相当或更高的基因转移。重要的是,集落形成祖细胞的克隆分析指示与本体cd34+靶标相比,当靶向cd34+/cd38-群体时转导更高百分比,而不是转导恒定分数但使用更高载体拷贝。理想地,临床应用将导致类似较高百分比的转导的hsc以获得更好功效,并且限制每个转导的细胞的vcn以获得更好安全性。
有趣的是,nsg小鼠中的体内移植研究证明与对应物cd34+/cd38-细胞相比,cd34+/cd38-细胞对植入的效力高大约100倍,具有基本上相等的植入贡献。不能从植入的nsg小鼠的骨髓获得足够的人细胞来基于其荧光标记分选出源自cd34+/cd38+细胞和cd34+/cd38-细胞的细胞,这样使得不能直接地测量绝对vcn/细胞来确定在cd34+/cd38-细胞的植入后代中是否存在如在体外所观察到的较高的载体拷贝。相反,用来自植入nsg小鼠的总骨髓细胞进行了ddpcr以定量用于标记cd34+/cd38-细胞或cd34+/cd38+细胞的特异性荧光报告基因,针对所述骨髓的人细胞含量进行标准化,并且指示1%cd34+/cd38-细胞与移植的99%cd34+/cd38-对造血的类似贡献。然而,有可能在骨髓中缺乏用于标记cd34+/cd38-细胞的载体的较高平均hsc可指示高vcn导致对转导的hsc的细胞毒性和对植入的降低的贡献。
总之,除了在此所示的对scd基因疗法的特定益处外,这些发现可应用于使用hsc的基因疗法的任何方法。基于在异种移植研究中的观察结果,在基因疗法中使用cd34+/cd38-细胞将允许使用较少量的载体来转导靶细胞,但仍然可产生足够的植入。这些发现与证明cd34+/cd38-细胞的良好植入能力的其他研究一致(case等人(1999)proc.natl.acad.sci.usa,96:2988-2993;geronimi等人(2003)stemcells,21:472-480;guenechea等人(2000)mol.ther.1:566-573;haas等人(2000)mol.ther.2:71-80)。
最近的公布描述了ldl受体作为用于常见地用于假型慢病毒载体的水疱性口炎病毒(vsv)的主要受体(finkelstein等人(2013)proc.natl.acad.sci.usa,110:7306-7311)。如果ldl受体表达在cd34+/cd38-细胞中较高,则存在更多载体结合至所述细胞、摄取至所述细胞中且最终整合至基因组中的可能性,从而导致更高的观察到的vcn。当对ldl受体的表达进行分析时,在未分级分离的cd34+和cd34+/cd38-细胞群体中,在用细胞因子刺激48小时(对应于转导时间)后存在很少静止的表达ldl受体的细胞和表达的相对等效的诱导。因此,用于vsv-g假型载体的受体的较高表达不太可能是增加的cd34+/cd38-细胞转导的机制。
当使用经rd-114逆转录病毒包膜蛋白假型化的ccl-βas3-fblv载体转导两个细胞群体时,再次存在cd34+/cd38-细胞相较于cd34+细胞的更高转导。这些结果加强了以下观察结果:尽管使用不同的包膜蛋白,对cd34+/cd38-细胞的转导的增加的敏感性(sandrin等人(2002)blood100:823-832;bell等人(2010)exp.biol.med.234:1269-1276;rasko等人(1999)proc.natl.acad.sci.usa,96:2129-2134)。
应了解,本文所述的实施例和实施方案仅用于说明性目的并且根据其产生的各种修改或变化将为本领域技术人员所想到并且欲包括于本申请的精神和权限以及随附权利要求的范围内。本文引用的所有公布、专利和专利申请出于所有目的据此以引用的方式整体并入本文。
- 还没有人留言评论。精彩留言会获得点赞!