噻吩三唑波拉(Bola)化合物的制作方法
本发明涉及多功能液晶材料领域,尤其是一种杂环波拉(Bola)两亲化合物,及其制备方法。
研究背景
近年来,杂环材料受到了人们广泛的关注。这些杂环材料在光电材料、有机薄膜晶体管、荧光传感器和有机太阳能电池等领域均有了广泛的应用。杂环也已经被广泛引入液晶中,实现了多种复杂的新型液晶相,例如:层列相、柱相以及三维立方相。这些杂环的引入也赋予这些液晶材料在光学、电学、生物学和药物学等领域拥有广泛的应用前景。虽然已经报道过噻吩Bola化合物,但仅限于对他们自组装性质的报道,这类化合物的推广和应用仍然为空白。三唑,噻二唑等杂环类化合物是当前功能材料的研究热点。因此将三唑和噻吩杂环同时引入Bola两亲化合物中有望获得新型的多功能材料,相关的研究未见文献报道。
技术实现要素:
本发明的目的是制备噻吩三唑Bola衍生物和获得多重功能性材料。将杂环引入Bola两亲化合物中实现了自组装性能,能应用于纳米光电材料的研究和包药领域、传感性能可应用于荧光探针、光学性能可应用于光学器件制备等。这些材料的多功能特性使其有望在现实生活中被广泛应用。
本发明的噻吩三唑波拉(Bola)化合物,其结构为:
所述的噻吩三唑波拉(Bola)化合物,其特征在于该化合物的制备步骤如下:
步骤1,将溴代烷烃和过量的镁屑反应所得的格氏试剂通过Kumada反应引入到3-溴噻吩或者3,4-二溴噻吩中,合成化合物3-烷基噻吩或者3,4-二烷基噻吩,该化合物称为2/n;
步骤2,化合物2/n在避光的条件下分批加入多于二倍量的NBS,使其取代成2,5-二溴-3-烷基噻吩或2,5-二溴-3,4-二烷基噻吩,该化合物称为3/n;
步骤3,将化合物3/n与化合物4-甲氧基苯硼酸在Suzuki条件下发生偶联反应得到化合物3-烷基-2,5-二(4-甲氧基苯基)噻吩或3,4-二烷基-2,5-二(4-甲氧基苯基)噻吩,该化合物称为4/n;
步骤4,将得到的中间体4/n在低温条件下去甲氧基化得到中间体2,5-二(4'-羟基苯基)-3-烷基噻吩或2,5-二(4'-羟基苯基)-3,4-二烷基噻吩,该化合物称为6/n;
步骤5,在N2保护下,将中间体5/n和溴丙炔反应得到中间体7/n;
步骤6,中间体7/n与4-叠氮基苯酚在Click的条件下发生反应生成中间体8/n;
步骤7,中间体8/n再与烯丙基溴反应得到中间体9/n;
步骤8,中间体9/n用OsO4的正丁醇溶液氧化分别得到最终产物TT1/n、TTB/n或TT2/n。
本发明的噻吩三唑波拉(Bola)化合物,具有液晶、凝胶、传感器及光学性能。
(1)液晶性能
所制备的噻吩三唑Bola衍生物TT1/n,TTB/n和TT2/n能自组装成多种柱相液晶,如下表一。带有较短烷基侧链的化合物TT1/n(n = 8 - 12)和TT2/n (n = 8 - 10)不具有液晶性质,而带有较长烷基侧链的化合物TT1/n(n = 14 - 18)和TT2/n (n = 12 - 16)能自组装成四方柱相(Colsqu/p4mm)。带有较短烷基侧链的化合物TTB/n(n = 4 – 6)不具有液晶性质,而带有较长烷基侧链的化合物TTB/n(n = 7 – 8)能自组装成由三角形构成的六方柱相(Colhex/p6mm)。可见化合物能形成有序的纳米液晶相结构,可以用为纳米光电材料的研究。
(2)凝胶性能
所制备的噻吩三唑Bola衍生物TT1/n,TTB/n和TT2/n均含极性三唑杂环和末端二醇基团,可形成氢键凝胶,如化合物TT1/16,在其不同溶剂中形成凝胶的能力做了系统的测试,如下表二所示。化合物TT1/16在极性较小的溶剂中不能溶解(如:正己烷、环己烷等);而在极性较大的溶剂中形成溶液(如:DMF,DMSO等);在二氯甲烷,乙酸乙酯,丙酮等溶剂中能形成白色的有机凝胶。通过SEM可以观察到这些白色凝胶是长度超过20 um,直径大约为188-217 nm的三维网络状结构。形成凝胶的性质使得这些化合物在包药领域具有良好的应用前景。
(3)光学性能
经过光谱测试,所制备的噻吩三唑Bola衍生物TT1/n,TTB/n和TT2/n在四氢呋喃(THF)溶液中的紫外最大吸收波长均在316-320 nm,化合物TT1/n,TTB/n和TT2/n在THF溶液中的荧光最大发射波长均在411-414 nm,最大发射波长位于蓝光区间,可作为蓝光材料,如表三所列。
(4)离子识别性能-铁离子化学传感器
据文献报道三唑类化合物可通过N原子与金属离子配位使荧光增强或者减弱,从而达到检测有害金属离子的作用。化合物TT1/n,TTB/n和TT2/n含有三唑杂环并且具有荧光性质,选定Mg2+, Ag+, Cd2+, Co2+, Cr2+, Cs+, Cu2+, Fe3+, Pb2+, Li+, Ni2+ 和 Hg2+金属离子测定化合物对金属离子的识别作用,分别将这些金属离子的水溶液(0.01 mmol/L)滴加到水和丙酮体积比为1 : 2的TTB/7溶液中(10-6 mol/L),测试结果表明,滴加Fe3+离子后,溶液会发生荧光淬灭,因此化合物TTB/7对Fe3+离子具有极好的专一性识别。为了确定识别Fe3+离子的最小浓度,做了定量分析,当Fe3+离子的浓度为该化合物浓度的10倍当量时,荧光强度不再发生变化,从而可确定检测Fe3+离子的检测限度为10-5 mol/L。化合物可作为荧光传感器用于检测活细胞中的Fe3+离子。
说明书附图
图1a是化合物TT1/14的液晶织构图;
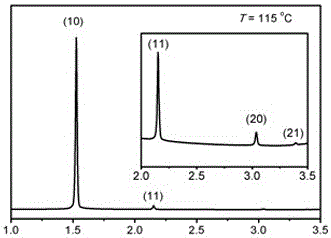
图1b是化合物TT1/14的XRD图;
图1c是化合物TT1/14的重建电子云密度图;
图1d是化合物TT1/14的分子自组装模型图;
图2a是化合物TTB/7的液晶织构图;
图2b是化合物TTB/7的XRD图;
图2c是化合物TTB/7的重建电子云密度图;
图2d是化合物TTB/7的分子自组装模型图;
图3a是TT1/16在丙酮中形成凝胶的SEM图a;
图3b是TT1/16在丙酮中形成凝胶的SEM图b;
图4是化合物TTB/7溶液对Fe3+离子的荧光强度变化图;
图5是化合物TTB/7溶液对Fe3+离子荧光强度改变图;
图6是向化合物TTB/7溶液中增加Fe3+离子浓度的荧光强度变化图;
图7是化合物TTB/7溶液中增加Fe3+离子浓度的滴定曲线图;
图8是噻吩三唑波拉(Bola)化合物的合成线路图。
具体实施方式
下面将结合附图进一步说明本发明的技术方案。
实施例1:噻吩三唑波拉(Bola)化合物TT1/n,通过以下的制备方法获得。
(i)3-烷基噻吩2a/n的合成
氮气保护下,在装有冷凝管、恒压滴液漏斗的两口烧瓶中,加入0.48 g镁屑(20.0 mmol),缓慢滴加 20 mL溶有10.0 mmol溴代烷烃的无水THF混合溶液,加完后在78 °C条件下回流反应1小时后冷至室温,移至另一恒压漏斗中备用。
另取一两口烧瓶,装置同上,加入1.00 g 3-溴噻吩 (约6.13 mmol)、10 mL THF、65 mg Ni(dppp)Cl2,在冰浴下缓慢滴加刚制得的格氏试剂,加完后继续反应1 h,然后移至油浴锅中,回流反应15 h。冷却至室温,蒸出THF,然后加入30 mL 冰冷的2 mol/L 的HCl,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,过滤,减压蒸馏蒸出溶剂,粗产品经过柱层析进行分离提纯(洗脱剂:石油醚,Rf = 0.8)。
(ii)2,5-二溴-3-烷基噻吩3a/n的合成
在干燥的单口烧瓶中,加入10.0 mmol 3-烷基噻吩2a/n和无水THF (20 mL),在避光条件下分批缓慢地加入22.0 mmol NBS,反应过夜。加入10 mL水停止反应,乙酸乙酯萃取三次,有机相依次用10% NaOH水溶液,水洗涤,无水Na2SO4干燥,减压蒸馏蒸出溶剂,粗产品经过柱层析进行分离提纯(洗脱剂:石油醚,;Rf = 0.9)。
(iii)3-甲氧基-2-5-二(4-甲氧基苯基)噻吩4a/n的合成
氮气保护下在装有冷凝管、恒压滴液漏斗的两口烧瓶中,加入2 mmol的化合物3a/n,4.8 mmol对甲氧基苯硼酸,THF:H2O = 1:1的混合溶液,并在超声除氧3-5 min,之后快速加入25 mg 四(三苯基磷)钯,加完后在78 °C条件下回流反应15 h。冷却至室温,减压蒸馏蒸出THF,然后加入30 mL水,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,减压蒸馏蒸出溶剂,粗产品经过柱层析进行分离提纯(洗脱剂:石油醚,Rf = 0.4)。
(iv) 化合物6a/n的合成
在-78 °C条件下,将1 mmol化合物4a/n溶于10-20 mL无水二氯甲烷,搅拌5-10 min后快速加入BBr3后反应15 h,恢复至室温,然后缓慢加入30 mL水,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,减压蒸馏蒸出溶剂,粗产品经过柱层析进行分离提纯(洗脱剂:石油醚:乙酸乙酯 = 5 : 1,Rf = 0.4)。
(v) 化合物7a/n的合成
在N2氛围下,在50 mL的两口圆底烧瓶中分别加入2.0 mmol 的化合物6a/n、6 mmol碳酸钾和5-10 mL乙腈,并在70 °C条件下回流5-10 min后快速加入6.5 mmol烯丙基溴反应15 h。冷却至室温,减压蒸馏蒸出乙腈,然后加入30 mL 水,乙酸乙酯萃取三次,有机层通过无水Na2SO4 干燥,减压蒸馏除出溶剂,粗产品通过柱层析进行分离提纯(洗脱剂:石油醚,Rf = 0.3)。
(vi) 化合物8a/n的合成
在50 ml的圆底烧瓶中分别加入1.5 mmol的化合物7a/n、 1.2 mmol的抗坏血酸钠、0.7 mmol的无水硫酸铜、叔丁醇: 水 = 1 : 1的溶液10 mL和10 mL THF和4 mmol 的4-叠氮基苯酚并在室温下反应15 h。减压蒸馏蒸出THF,然后加入30 mL水,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,减压蒸馏蒸出溶剂,粗产品通过柱层析进行分离提纯(洗脱剂:乙酸乙酯 : 石油醚 = 3 : 1,Rf = 0.2)。
(vii) 化合物9a/n的合成
在50 mL的圆底烧瓶中分别加入1.0 mmol化合物8a/n、3 mmol碳酸钾和15mL乙腈并在75 °C条件下回流5-10 min,之后快速加入2.5 mmol的烯丙基溴,反应5 h。冷却至室温后蒸出乙腈,然后加入30 mL 水,乙酸乙酯萃取三次,有机层通过无水Na2SO4 干燥,减压蒸馏蒸出溶剂,粗产品通过柱层析进行分离提纯(洗脱剂:石油醚 : 乙酸乙酯 = 2 : 1,Rf = 0.3)。
(viii) 化合物TT1/n的合成
在50 mL的圆底烧瓶中分别加入1.0 mmol化合物9a/n、1 mL OsO4的正丁醇溶液(0.004 mol/L)、5-10 mL丙酮和NMMNO的水溶液1 mL,在50 °C条件下回流5 h。冷却至室温后加入饱和的NaS2O3水溶液,并在室温下搅拌30分钟。然后加入30 mL水,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,减压蒸馏除蒸出溶剂,粗产品通过柱层析进行分离(洗脱剂:乙酸乙酯,Rf = 0.2)。
实施例2:噻吩三唑波拉(Bola)化合物TTB/n, 通过以下的制备方法获得。
(i)3-烷基噻吩2b/n的合成
氮气保护下,在装有冷凝管、恒压滴液漏斗的两口烧瓶中,加入0.48 g镁屑(20.0 mmol),缓慢滴加 20 mL溶有10.0 mmol分枝溴代烷烃的无水THF混合溶液,加完后在78 °C条件下回流反应1小时后冷至室温,移至另一恒压漏斗中备用。
另取一两口烧瓶,装置同上,加入1.00 g 3-溴噻吩 (约6.13 mmol)、10 mL THF、65 mg Ni(dppp)Cl2,在冰浴下缓慢滴加刚制得的格氏试剂,加完后继续反应1 h,然后移至油浴锅中,回流反应15 h。冷却至室温,蒸出THF,然后加入30 mL 冰冷的2 mol/L 的HCl,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,过滤,减压蒸馏蒸出溶剂,粗产品经过柱层析进行分离提纯(洗脱剂:石油醚,Rf = 0.8)。
(ii)2,5-二溴-3-烷基噻吩3b/n的合成
在干燥的单口烧瓶中,加入10.0 mmol 3-烷基噻吩3b/n和无水THF (20 mL),在避光条件下分批缓慢地加入22.0 mmol NBS,反应过夜。加入10 mL水停止反应,乙酸乙酯萃取三次,有机相依次用10% NaOH水溶液,水洗涤,无水Na2SO4干燥,减压蒸馏蒸出溶剂,粗产品经过柱层析进行分离提纯(洗脱剂:石油醚,;Rf = 0.9)。
(iii)3-甲氧基-2-5-二(4-甲氧基苯基)噻吩4b/n的合成
氮气保护下在装有冷凝管、恒压滴液漏斗的两口烧瓶中,加入2 mmol的化合物3b/n,4.8 mmol对甲氧基苯硼酸,THF:H2O = 1:1的混合溶液,并在超声除氧3-5 min,之后快速加入25 mg 四(三苯基磷)钯,加完后在78 °C条件下回流反应15 h。冷却至室温,减压蒸馏蒸出THF,然后加入30 mL水,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,减压蒸馏蒸出溶剂,粗产品经过柱层析进行分离提纯(洗脱剂:石油醚,Rf = 0.4)。
(iv) 化合物6b/n的合成
在-78 °C条件下,将1 mmol化合物4b/n溶于10-20 mL无水二氯甲烷,搅拌5-10 min后快速加入BBr3后反应15 h,恢复至室温,然后缓慢加入30 mL水,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,减压蒸馏蒸出溶剂,粗产品经过柱层析进行分离提纯(洗脱剂:石油醚:乙酸乙酯 = 5 : 1,Rf = 0.4)。
(v) 化合物7b/n的合成
在N2氛围下,在50 mL的两口圆底烧瓶中分别加入2.0 mmol 的化合物6b/n、6 mmol碳酸钾和5-10 mL乙腈,并在70 °C条件下回流5-10 min后快速加入6.5 mmol烯丙基溴反应15 h。冷却至室温,减压蒸馏蒸出乙腈,然后加入30 mL 水,乙酸乙酯萃取三次,有机层通过无水Na2SO4 干燥,减压蒸馏除出溶剂,粗产品通过柱层析进行分离提纯(洗脱剂:石油醚,Rf = 0.3)。
(vi) 化合物8b/n的合成
在50 ml的圆底烧瓶中分别加入1.5 mmol的化合物7b/n、 1.2 mmol的抗坏血酸钠、0.7 mmol的无水硫酸铜、叔丁醇: 水 = 1 : 1的溶液10 mL和10 mL THF和4 mmol 的4-叠氮基苯酚并在室温下反应15 h。减压蒸馏蒸出THF,然后加入30 mL水,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,减压蒸馏蒸出溶剂,粗产品通过柱层析进行分离提纯(洗脱剂:乙酸乙酯 : 石油醚 = 3 : 1,Rf = 0.2)。
(vii) 化合物9b/n的合成
在50 mL的圆底烧瓶中分别加入1.0 mmol化合物8b/n、3 mmol碳酸钾和15mL乙腈并在75 °C条件下回流5-10 min,之后快速加入2.5 mmol的烯丙基溴,反应5 h。冷却至室温后蒸出乙腈,然后加入30 mL 水,乙酸乙酯萃取三次,有机层通过无水Na2SO4 干燥,减压蒸馏蒸出溶剂,粗产品通过柱层析进行分离提纯(洗脱剂:石油醚 : 乙酸乙酯 = 2 : 1,Rf = 0.3)。
(viii) 化合物TTB/n的合成
在50 mL的圆底烧瓶中分别加入1.0 mmol化合物9b/n、1 mL OsO4的正丁醇溶液(0.004 mol/L)、5-10 mL丙酮和NMMNO的水溶液1 mL,在50 °C条件下回流5 h。冷却至室温后加入饱和的NaS2O3水溶液,并在室温下搅拌30分钟。然后加入30 mL水,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,减压蒸馏除蒸出溶剂,粗产品通过柱层析进行分离(洗脱剂:乙酸乙酯,Rf = 0.2)。
实施例3:噻吩三唑波拉(Bola)化合物TT2/n,通过以下的制备方法获得。
(i)3,4-二烷基噻吩2c/n的合成
氮气保护下,在装有冷凝管、恒压滴液漏斗的两口烧瓶中,加入0.48 g镁屑(20.0 mmol),缓慢滴加 20 mL溶有10.0 mmol溴代烷烃的无水THF混合溶液,加完后在78 °C条件下回流反应1小时后冷至室温,移至另一恒压漏斗中备用。
另取一两口烧瓶,装置同上,加入0.9 g 3,4-二溴噻吩 (约3.72 mmol),10 mL THF,65 mg Ni(dppp)Cl2,在冰浴下缓慢滴加刚制得的格氏试剂,加完后继续反应1 h,然后移至油浴锅中,回流反应15 h。冷却至室温,蒸出THF,然后加入30 mL 冰冷的2 mol/L 的HCl,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,过滤,减压蒸馏蒸出溶剂,粗产品经过柱层析进行分离提纯(洗脱剂:石油醚,Rf = 0.8)。
(ii)2,5-二溴-3,4-二烷基噻吩3c/n的合成
在干燥的单口烧瓶中,加入3,4-二烷基噻吩和无水THF (20 mL),在避光条件下分批缓慢地加入22.0 mmol NBS,反应过夜。加入10 mL水停止反应,乙酸乙酯萃取三次,有机相依次用10% NaOH水溶液,水洗涤,无水Na2SO4干燥,减压蒸馏蒸出溶剂,粗产品经过柱层析进行分离提纯(洗脱剂:石油醚,;Rf = 0.9)。
(iii)3,4-二甲氧基-2-5-二(4-甲氧基苯基)噻吩4c/n的合成
氮气保护下在装有冷凝管、恒压滴液漏斗的两口烧瓶中,加入2 mmol的化合物3c/n,4.8 mmol对甲氧基苯硼酸,THF:H2O = 1:1的混合溶液,并在超声除氧3-5 min,之后快速加入25 mg 四(三苯基磷)钯,加完后在78 °C条件下回流反应15 h。冷却至室温,减压蒸馏蒸出THF,然后加入30 mL水,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,减压蒸馏蒸出溶剂,粗产品经过柱层析进行分离提纯(洗脱剂:石油醚,Rf = 0.4)。
(iv) 化合物6c/n的合成
在-78 °C条件下,将1 mmol化合物4c/n溶于10-20 mL无水二氯甲烷,搅拌5-10 min后快速加入BBr3后反应15 h,恢复至室温,然后缓慢加入30 mL水,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,减压蒸馏蒸出溶剂,粗产品经过柱层析进行分离提纯(洗脱剂:石油醚:乙酸乙酯 = 5 : 1,Rf = 0.4)。
(v) 化合物7c/n的合成
在N2氛围下,在50 mL的两口圆底烧瓶中分别加入2.0 mmol 的化合物6c/n、6 mmol碳酸钾和5-10 mL乙腈,并在70 °C条件下回流5-10 min后快速加入6.5 mmol烯丙基溴反应15 h。冷却至室温,减压蒸馏蒸出乙腈,然后加入30 mL 水,乙酸乙酯萃取三次,有机层通过无水Na2SO4 干燥,减压蒸馏除出溶剂,粗产品通过柱层析进行分离提纯(洗脱剂:石油醚,Rf = 0.3)。
(vi) 化合物8c/n的合成
在50 ml的圆底烧瓶中分别加入1.5 mmol的化合物7c/n、1.2 mmol的抗坏血酸钠、0.7 mmol的无水硫酸铜、叔丁醇: 水 = 1 : 1的溶液10 mL和10 mL THF和4 mmol 的4-叠氮基苯酚并在室温下反应15 h。减压蒸馏蒸出THF,然后加入30 mL水,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,减压蒸馏蒸出溶剂,粗产品通过柱层析进行分离提纯(洗脱剂:乙酸乙酯 : 石油醚 = 3 : 1,Rf = 0.2)。
(vii) 化合物9c/n的合成
在50 mL的圆底烧瓶中分别加入1.0 mmol化合物8c/n、3 mmol碳酸钾和15mL乙腈并在75 °C条件下回流5-10 min,之后快速加入2.5 mmol的烯丙基溴,反应5 h。冷却至室温后蒸出乙腈,然后加入30 mL水,乙酸乙酯萃取三次,有机层通过无水Na2SO4 干燥,减压蒸馏蒸出溶剂,粗产品通过柱层析进行分离提纯(洗脱剂:石油醚 : 乙酸乙酯 = 2 : 1,Rf = 0.3)。
(viii) 化合物TT2/n的合成
在50 mL的圆底烧瓶中分别加入1.0 mmol化合物9c/n、1 mL OsO4的正丁醇溶液(0.004 mol/L)、5-10 mL丙酮和NMMNO的水溶液1 mL,在50 °C条件下回流5 h。冷却至室温后加入饱和的NaS2O3水溶液,并在室温下搅拌30分钟。然后加入30 mL水,乙酸乙酯萃取三次,有机层通过无水Na2SO4干燥,减压蒸馏除蒸出溶剂,粗产品通过柱层析进行分离(洗脱剂:乙酸乙酯,Rf = 0.2)。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种紫外线吸收剂uv-234的制备方法
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑5-甲基间苯二甲酸锰配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑镉混配配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑间苯二甲酸镉配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑锌混配配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑三维锌配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑对苯二甲酸铜配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-双三唑均苯四酸铜配合物单晶与应用
- 一种4-苯基-3-((4,6-二甲基嘧啶-2-基硫代)甲基)-5-苄硫基三唑类化合物及其应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑二维铜配合物单晶与应用