一种抗结核杆菌化合物及其制备方法与应用与流程
本发明涉及一种抗结核杆菌化合物及其制备方法与应用。
背景技术:
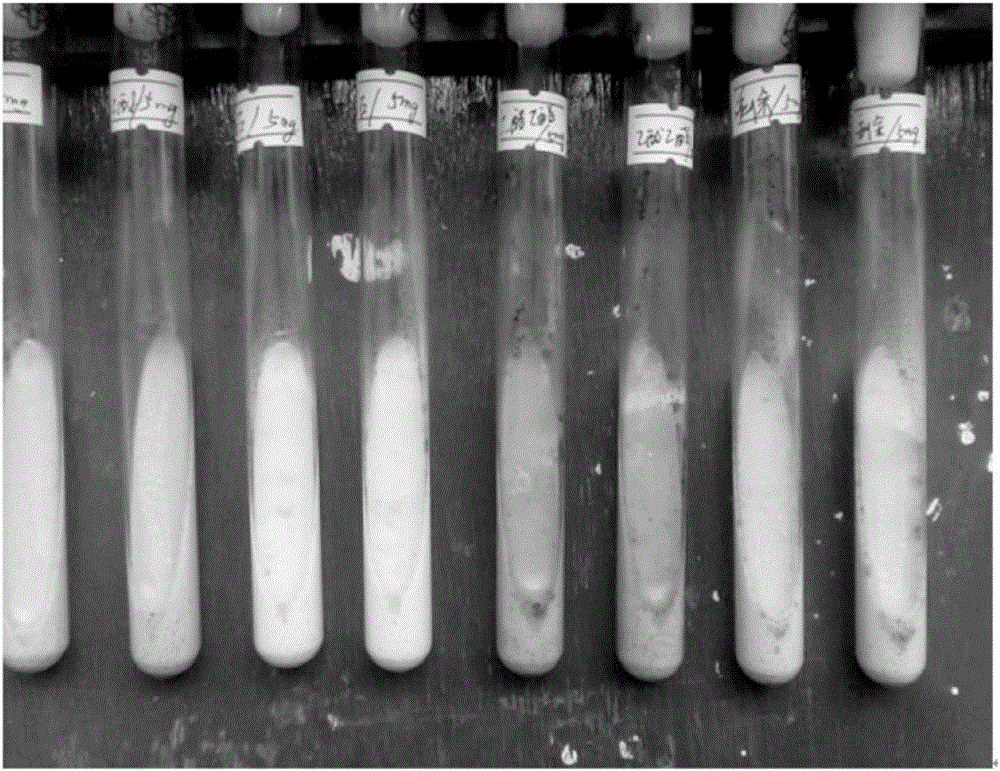
:结核病(Tuberculosis,TB)是由结核分枝杆菌(MycobacteriumTuberculosis,MTB)引起的一种慢性致死性疾病,是危害人类健康和导致人类死亡的重大传染性疾病,其致死率仅次于艾滋病(Ac-quiredImmunodeficiencysyndrome,AIDS)。由于抗结核药物的大量使用和抗结核作用位点的突变,导致耐多药结核病(multidrug-resistanttuberculosis,MDR-TB)不断出现,再加上近年来患有艾滋病人口数量的增长,使结核病与人类免疫缺陷病毒(humanimmunodeficiencyvirus,HIV)并发发病率急剧增加,这些问题的出现都给结核病的治疗带来巨大的挑战,这无疑说明结核病已经成为人类健康的重大威胁,亟待解决。技术实现要素:本发明的目的是提供一种抗菌化合物及其制备方法与应用。本发明所提供的化合物如式Ⅰ所示。所述化合物在制备抗结核杆菌药物中的应用也属于本发明的保护范围。本发明的再一个目的是提供一种抗菌药物。本发明所提供的抗菌药物的活性成分为以上式Ⅰ所示的化合物。在本发明中,所述抗菌药物为抗结核杆菌药物。本发明的另一个目的是提供所述化合物的制备方法。本发明所提供的所述化合物的制备方法,是对独山瓜馥木干燥根进行甲醇回流提取,获得独山瓜馥木甲醇浸膏,再对所得甲醇浸膏进行分离和纯化,从而得到所述化合物。进一步,所述甲醇浸膏是按照如下方法制备得到的:将所述独山瓜馥木干燥根切块,加入2倍重量的甲醇,回流提取3次,每次1.5h,去除甲醇(如用旋转蒸发仪减压回收),水浴蒸干,获得所述独山瓜馥木甲醇提取物。在上述方法中,所述分离和纯化的方法具体可包括如下步骤:(a)向所述甲醇提取物中加水,用可溶解所述化合物的有机溶剂进行萃取,收集有机相,去除有机溶剂,获得含有权利要求1所述化合物的萃取物;(b)将所述萃取物依次经过所述硅胶柱层析A、硅胶柱层析B、制备薄层色谱,分离得到以上式Ⅰ所示的化合物。在步骤(a)中,所述可溶解所述化合物的溶剂可为乙酸乙酯。步骤(b)中,进行所述硅胶柱层析A时,采用的硅胶粒度为200-300目,洗脱体积为5500mL,洗脱体积的第1-500mL采用的洗脱液由体积比为10:1的石油醚和乙酸乙酯混合而成,洗脱体积的第501-1000mL采用的洗脱液由体积比为10:2的石油醚和乙酸乙酯混合而成,洗脱体积的第1001-1500mL采用的洗脱液由体积比为10:3的石油醚和乙酸乙酯混合而成,洗脱体积的1501-2000mL采用的洗脱液由体积比为10:4的石油醚和乙酸乙酯混合而成,洗脱体积的2001-2500mL采用的洗脱液由体积比为10:5的石油醚和乙酸乙酯混合而成,洗脱体积的2501-3000mL采用的洗脱液由体积比为10:6的石油醚和乙酸乙酯混合而成,洗脱体积的第3011-3500mL采用的洗脱液由体积比为10:7的石油醚和乙酸乙酯混合而成,洗脱体积的第3501-4000mL采用的洗脱液由体积比为10:8的石油醚和乙酸乙酯混合而成,洗脱体积的第4001-4500mL采用的洗脱液由体积比为10:9的石油醚和乙酸乙酯混合而成,洗脱体积的第4501-5000mL采用的洗脱液由体积比为10:10的石油醚和乙酸乙酯混合而成,洗脱体积的第5001-5500mL采用的洗脱液乙酸乙酯洗脱而成。在步骤(b)中,进行所述硅胶柱层析B时,采用的硅胶粒度可为200-300目,洗脱液由体积比为10:9的石油醚和乙酸乙酷混合而成,洗脱体积1000mL。进行所述制备薄层色谱时,采用的吸附剂为硅胶,硅胶粒度可为200-400目,展开剂由体积比为24:1的二氯甲烷和甲醇混合而成。在本发明的一个实施例中,进行所述制备薄层色谱时,采用的硅胶为GF254型号的硅胶板(购自青岛海洋化工)。在上述方法中,步骤(a)可为:向所述甲醇提取物中加水,用石油醚进行萃取,收集水相;再用乙酸乙酯对所述水相进行萃取,收集有机相,即获得所述“含有所述化合物的萃取液”。在本发明的一个实施例中,步骤(a)更加具体为:向所述甲醇提取物中加入1倍重量的水,搅拌得到水悬液;再向所述水悬液中加入等体积的石油醚,萃取30分钟,萃取三次,收集水相;再向所述水相中加入等体积的乙酸乙酯,萃取30分钟,萃取三次,收集有机相,去除有机溶剂,蒸干即获得所述“含有所述化合物的萃取物”。在上述方法中,步骤(b)具体可为:将所述萃取物进行所述硅胶柱层析A,收集中间产物Fra.I,所述中间产物Fra.I在以体积比为10:9的石油醚和乙酸乙酯混合液作为展开剂,以粒度为100-200目的硅胶作为固定相的薄层层析(操作温度27℃)中,比移值(Rf)为0.30-0.42;再将所述中间产物Fra.I进行所述硅胶柱层析B,收集中间产物Subfra.I3,所述中间产物Subfra.I3在以体积比为10:9的石油醚和乙酸乙酯的混合液作为展开剂,以粒度为200-300目的硅胶作为固定相的薄层层析(操作温度27℃)中,比移值(Rf)为0.28-0.35;再将所述中间产物Subfra.I3进行所述制备薄层色谱,收集比移值(Rf)为0.27的样品,获得以上式Ⅰ所示的化合物。其中,所述中间产物Fra.I为在进行所述硅胶柱层析A时,以体积比为10:9的石油醚和乙酸乙酯混合液作为洗脱液进行洗脱所得,对应洗脱体积的4001-4500mL。所述中间产物Subfra.I3为在进行所述硅胶柱层析B时,以体积比为10:9的石油醚和乙酸乙酯的混合液作为洗脱液进行洗脱所得,对应洗脱体积的401-600mL。更加具体的,步骤(b)中,在进行所述硅胶柱层析A时,采用的层析柱的规格为60X900mm,柱温为27℃,所用硅胶为1.8kg,上样量为36g所述萃取物,流速为*ml/min,在进行所述硅胶柱层析B时,采用的层析柱的规格为20X900mm,柱温为27℃,所用硅胶为900g,上样量为1.5g所述中间产物Fra.I,流速为*ml/min。在进行所述制备薄层色谱时,采用的薄层板规格为20X20cm,操作温度27℃;上样量0.5ml;展开时间1h。实验证明,本发明提供的化合物结构全新,具有较好的抗结核杆菌活性,适宜于抗菌先导化合物研究或制备抗菌药物。本发明所用的天然产物活性成分制备方法选择了能产生具有优异抗菌活性化合物的独山瓜馥木(Fissistigmacavaleriei),提取方法成熟,工艺简便,所得产物产率高,经核磁共振检测,其结构正确。附图说明图1为本发明所述TLC图;图2位本发明所述活性测试图;附图说明:图1从左到右依次为乙醇提取物、石油醚萃取物、乙酸乙酯萃取物和萃取后剩余物;图2从左至右依次为:乙醇提取物、石油醚萃取物、乙酸乙酯萃取物、萃取后剩余物。具体实施方式下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。独山瓜馥木干燥根(Fissistigmacavaleriei):采自贵州省黔东南州独山县。独山瓜馥木干燥根(Fissistigmacavaleriei)凭证标本已于2011年04月03日保藏于贵州大学药学院。当然,也可从商业途径购得独山瓜馥木干燥根(Fissistigmacavaleriei)。结核分枝杆菌(MycobacteriumTuberculosis,MTB)为H37Rv标准株。实施例1抗菌化合物的制备及鉴定所用试剂均为分析纯(天津市致远化学试剂有限公司);硅胶柱色谱所用硅胶100-200目、200-300目均购自青岛海洋化工厂;制备薄层色谱所用硅胶GF254购自青岛市青岛海洋化工分厂。一、抗菌化合物的制备1、独山瓜馥木干燥根3Kg,切块,分批次加入适量甲醇,加热回流提取3次,每次1.5h,用旋转蒸发仪减压回收溶剂,从而去除甲醇,获得独山瓜馥木甲醇提取物。独山瓜馥木干燥根乙酸乙酯萃取物的获得2、向步骤1所得的独山瓜馥木根甲醇提取物中加入1倍重量的水,搅拌使其混悬,再向其中加入等体积的石油醚,萃取0.3h,萃取三次,收集水相;再向所述水相中加入等体积的乙酸乙酯,萃取0.5h,萃取三次,收集有机相,用旋转蒸发仪减压回收溶剂,从而去除乙酸乙酯,获得独山瓜馥木根乙酸乙酯萃取物。3、独山瓜馥木乙酸乙酯萃取物的硅胶柱层析硅胶柱层析条件如下硅胶柱(常压柱)规格:60X900mm(海门市华凯实验玻璃仪器有限公司);柱温为27℃;所用硅胶(200-300目)量为1.8kg;独山瓜馥木根乙酸乙酯萃取物的上样量为36g;洗脱液及洗脱程序如表1所示;流速为2ml/min;连续收集洗脱液,每100mL收集一管。表1独山瓜馥木根乙酸乙酯萃取物的硅胶柱层析的洗脱程序洗脱液编号洗脱液成分石油醚:乙酸乙酯洗脱体积(ml)洗脱液1体积比10:11-500洗脱液2体积比10:2501-1000洗脱液3体积比10:31001-1500洗脱液4体积比10:41501-2000洗脱液5体积比10:52001-2500洗脱液6体积比10:62501-3000洗脱液7体积比10:73001-3500洗脱液8体积比10:83501-4000洗脱液9体积比10:94001-4500洗脱液10体积比10:104501-5000洗脱液11体积比0:15001-5500对所收集的各管洗脱液进行薄层层析(TLC)检测,以各管洗脱液进行硅胶柱层析洗脱时采用的相应组分的洗脱液作为展开剂,以粒度为100-200目的硅胶作为固定相,操作温度为27℃。计算10种洗脱液对应的10个流份的比移值(Rf)。结果显示:将该步骤所收集的10个流份,分别记为Fra.A,Fra.B,Fra.C,Fra.D,Fra.E,Fra.F,Fra.G,Fra.H,Fra.I,Fra.J,Fra.K。其中,Fra.A为用洗脱液石油醚:乙酸乙酯(体积比10:1)洗脱得到的(对应洗脱体积的第1-500ml),比移值(Rf)为0.55-0.70;Fra.B为用洗脱液石油醚:乙酸乙酯(体积比10:2)洗脱得到的(对应洗脱体积的第501-1000ml),比移值(Rf)为0.50-0.67;Fra.C为用洗脱液石油醚:乙酸乙酯(体积比10:3)洗脱得到的(对应洗脱体积的第1001-1500ml),比移值(Rf)为0.5-0.60;Fra.D为用洗脱液石油醚:乙酸乙酯(体积比10:4)洗脱得到的(对应洗脱体积的第1501-2000ml),比移值(Rf)为0.45-0.62;Fra.E为用洗脱液石油醚:乙酸乙酯(体积比10:5)洗脱得到的(对应洗脱体积的第2001-2500ml),比移值(Rf)为0.40-0.58;Fra.F为用洗脱液石油醚:乙酸乙酯(体积比10:6)洗脱得到的(对应洗脱体积的2501-3000mL),比移值(Rf)为0.37-0.55;Fra.G为用洗脱液石油醚:乙酸乙酯(体积比10:7)洗脱得到的(对应洗脱体积的第3001-3500ml),比移值(Rf)为0.35-0.55;Fra.H为用洗脱液石油醚:乙酸乙酯(体积比10:8)洗脱得到的(对应洗脱体积的第3501-4000ml),比移值(Rf)为0.33-0.53;Fra.I为用洗脱液石油醚:乙酸乙酯(体积比10:9)洗脱得到的(对应洗脱体积的第4001-4500ml),比移值(Rf)为0.30-0.42;Fra.J为用洗脱液石油醚:乙酸乙酯(体积比10:10)洗脱得到的(对应洗脱体积的第4501-5000ml),比移值(Rf)为0.26-0.40。Fra.K为用洗脱液为乙酸乙酯洗脱得到的(对应体积5001-5500ml),比移值0.35-0.67。4,流份Fra.I的硅胶柱层析。硅胶柱(常压柱)规格:20X900mm(海门市华凯实验玻璃仪器有限公司);柱温为27℃;所用硅胶(200-300目)量为900g;上样量为1.5g(1.5g流份Fra.I蒸干溶剂后的固体);洗脱液为石油醚:乙酸乙酯(体积比10:9);流速为*ml/min;洗脱体积为1000ml。连续收集洗脱液,每20ml收集一管。洗脱体积的第1-200mL为第1个流份、洗脱体积的第201-400ml为第2个流份、洗脱体积的第401-600ml为第3个流份、洗脱体积的第601-800ml为第4个流份、洗脱体积的第801-1000ml为第5个流份。对所收集的各管洗脱液进行薄层层析(TLC)检测,用进行以上硅胶柱层析时采用的洗脱液(体积比为10:9的石油醚:乙酸乙酯)作为展开剂,以粒度为200-300目的硅胶作为固定相,操作温度为27℃。计算5个流份的比移值(Rf)。结果显示,将该步骤所收集的5个流份,分别记为Subfra.Il,Subfra.I2,Subfra.I3,Subfra.I4,Subfra.I5。其中,Subfra.Il对应洗脱体积的第1-200mL,比移值(Rf)为0.38-0.50;Subfra.I2对应洗脱体积的第201-400mL,比移值(Rf)为0.35-0.47;Subfra.I3对应洗脱体积的第401-600m,比移值(Rf)为0.32-0.40;Subfra.I4对应洗脱体积的第601-800mL,比移值为0.26-0.34,Subfra.I5对应洗脱体积的第801-1000ml,比移值为0.20-0.30。5,流份Subfra.I3的制备色谱薄层板规格:20X20cm(制备薄层色谱所用硅胶GF254购自烟台市化学工业研究所);操作温度27℃;上样量0.5ml;展开时间1h;展开剂为体积比为24:1的二氯甲烷:甲醇。上样展开完毕,进行显色,将检测到的样品连同硅胶一齐刮下,使用洗脱液(体积比为24:1的二氯甲烷:甲醇)将组分洗脱,将洗脱液蒸干。共得到40mg式Ⅰ化合物,在以上制备薄层色谱中的比移值(Rf)为0.5。二、抗菌化合物式Ⅰ的结构鉴定对步骤一得到的式Ⅰ化合物进行鉴定(1)外观:式Ⅰ化合物为黄色或淡黄色液体至固体。(2)溶解性:易溶于乙酸乙酯、二氯甲烷、甲醇等有机溶剂。(3)核磁共振谱:图1是式Ⅰ化合物的1H-NMR谱图,图2是式Ⅰ化合物的13C-NMR谱图。根据化合物的1H-NMR,13C-NMR,对两个化合物的核磁共振谱进行了研究并对13C信号进行了归属,见表2。并最终确定结构如下:式Ⅰ化合物氢谱和碳谱峰的归属,ES-MSm/z126[M]+;H-NMR(CDCl3)δ:4.9(-OH),4.7(6-H),6.5(4-H),7.3(3-H),9.5(CHO);13C-NMR(CDCl3)δ::57.0(6-C),161.5(5-C),111.6(4-C),124.0(3-C),152.0(2-C),178.1(CO)。注:溶剂为二氯甲烷,式Ⅰ化合物的NMR测试采用里瓦安核磁共振仪器(400兆赫兹),测试的溶剂均为氘代二氯甲烷。实施例2,式Ⅰ化合物的抗结核杆菌活性测定一、实验方法抗结核杆菌活性测定结核杆菌在改良罗氏培养基上生长良好,通过观察其在培养基上生长菌落的增殖情况来衡量式Ⅰ化合物的抗菌活性,具体操作如下。将结核杆菌H37Rv接种到含有改良罗氏培养基(上海博微生物科技有限公司)的固体斜面上,药物浓度分别为500μg/ml、2500μg/ml、5000μg/ml,阳性对照组为异烟肼2μg/ml,阴性对照组为5%二甲基亚砜,置于37℃的恒温培养箱中培养10天,统计待测化合物对结核杆菌的最低抑菌浓度值(MIC),通过观察结核杆菌的生长情况,生长被抑制80%以上的化合物浓度即为该化合物对结核杆菌的最低抑菌浓度值。实验重复3次。二、实验结果结果显示,在抗结核杆菌实验中,实施例1制备得到的式Ⅰ化合物对结核杆菌的抗菌活性MIC确定为500μg/ml,具体结果如表3所示。表3,式Ⅰ化合物的抗菌活性表3抗结核杆菌活性试验当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1