一种微反应器制备拉科酰胺的方法与流程
本发明涉及一种微反应器制备拉科酰胺的方法,属于药物合成技术领域。
背景技术:
拉科酰胺(Lacosamide),中文化学名:(R)-2-乙酰胺基-N-苄基-3-甲氧基丙酰胺,结构式如下:
拉科酰胺(Lacosamide)是比利时UCB医药有限公司开发的第三代治疗癫痫和神经性疼痛的药物。拉科酰胺是近三年来首个治疗癫痫部分发作的抗癫痫新药,对未控制的癫痫部分发作患者提供了治疗新手段。拉科酰胺是一种新型的N-甲基-D-天门冬氨酸(NMDA)受体甘氨酸位点结合拮抗剂,属于新型功能性氨基酸抗惊厥药物,具有抗惊厥和镇痛的双重作用。该药已于2008年9月和2008年10月分别获得欧盟医药管理局(EMEA)和美国食品药物管理局(FDA)的批准上市,商品名Vimpat,适用于17岁及以上伴或不伴有继发性全身性发作的部分性发作癫痫患者的辅助治疗。本品尚未在中国上市。关于拉科酰胺的合成方法有许多的文献介绍。
美国专利US6048899中报道了两条合成路线:
路线一:D-丝氨酸的氨基经过保护然后与苄胺缩合,经过碘甲烷甲基化,Pd-C还原,最后乙酰化得到拉科酰胺,化学反应方程式如下所示。
注:Cbz-为苄氧羰基。
该路线甲基化使用了碘甲烷、氧化银,价格昂贵;脱氨基保护使用了Pd-C还原,生产成本高,不利于工业化生产。
路线二:D-丝氨酸经氨基保护,碘甲烷甲基化后与苄胺缩合,Pd-C还原,最后乙酰化得到拉科酰胺,化学反应方程式如下所示。
注:Cbz-为苄氧羰基。
该路线同路线一相比,改变了甲基化以及缩合反应的顺序,步骤延长,但是也使用了甲基化碘甲烷、氧化银,价格昂贵;脱氨基保护也使用了Pd-C还原,生产成本高,不利于工业化生产。
专利US20080027137对以上路线中的氨基保护以及甲基化试剂进行了改进,具体反应方程式如下:
注Boc-为二叔丁氧羰基。
该方法氨基保护使用了二叔丁氧羰基代替苄氧羰基,避免了Pd-C的使用,操作简单,成本降低,但是仍然存在产品纯度及收率不高的问题。
专利CN103113256A公开的拉科酰胺的合成方法为:以D-丝氨酸为原料先经乙酰化、苄胺缩合、最后甲基化得到产品;专利CN102209707A公开的拉科酰胺的合成方法为:以2-溴-3-甲氧基丙酸为原料,经氯甲酸乙酯活化后与苄胺缩合,再与氨水发生取代反应,再对氨基乙酰化,然后经模拟移动床色谱(SMB)进行异构体的分离,得到的R构型的对映体即为拉科酰胺,S构型的对映体经消旋处理后得到外消旋的,再经分离后得到R构型的拉科酰胺(手性纯度达到99%),如此循环,理论上可以完全转化为R构型的拉科酰胺。该路线没有使用到甲基化试剂,同时也省去了氨基的保护这一操作,循环拆分的方法也较为新颖,提高了生产收率。但工艺较繁琐,原料价格较贵,成本高。
综上,目前的合成方法主要以D-丝氨酸为起始原料,反应过程中需要经过氨基保护和脱保护两步反应,进而造成步骤繁琐,产品纯度及收率不高的问题。
技术实现要素:
本发明克服了上述现有技术的不足,提供了一种微反应器制备拉科酰胺的方法。该方法采用微反应器进行反应,利用微反应器的高选择性避免了D-丝氨酸中的裸露氨基的甲基化副产物的产生,从而省略了传统方法中的氨基保护和脱保护两步反应,由5步反应减缩为3步反应,大大简化了反应步骤,提高了产品的收率。
本发明的技术方案是:一种微反应器制备拉科酰胺的方法,其特征是,
1)以D-丝氨酸为起始原料,与苯胺进行缩合反应得到R-2-氨基-N-苄基-3-羟基丙酰胺(化合物Ⅰ);
2)R-2-氨基-N-苄基-3-羟基丙酰胺再与甲基化试剂反应得到R-2-氨基-N-苄基-3-甲氧基丙酰胺(化合物Ⅱ);
3)然后与乙酰化试剂进行乙酰化反应得到拉科酰胺;
所述至少步骤2)在微反应器内进行反应。优选的,所述步骤2)和3)都在微反应器内进行反应。
优选的,所述甲基化试剂为硫酸二甲酯,所述乙酰化试剂为乙酸酐。
优选的,所述步骤1)的反应中加入叔胺和氯甲酸烷基酯。叔胺可以是二异丙基乙胺、N-甲基吗啉、三正丁胺、三乙胺、2,6-二甲基吡啶等,优先选择三乙胺。所述氯甲酸烷基酯可以是氯甲酸甲酯、氯甲酸乙酯、氯甲酸异丙酯、氯甲酸异丁酯等,优先选择氯甲酸异丁酯。
优选的,所述步骤2)在碱性条件下进行,所述碱可以是氢氧化钠、氢氧化钾、碳酸氢钠、碳酸氢钾等,优先选择氢氧化钠。
反应方程式如下所示。
注:IBCF为氯甲酸异丁酯;TEA为三乙胺。
本发明的制备方法,具体如下:
1)控制反应温度-15℃~0℃,将D-丝氨酸加入有机溶剂中,依次加入叔胺、氯甲酸烷基酯和苄胺进行反应至反应完全,得到R-2-氨基-N-苄基-3-羟基丙酰胺溶液(简称羟基丙酰胺溶液);所述D-丝氨酸、氯甲酸烷基酯、叔胺和苄胺的摩尔比为1:0.98~1.02:1.1~1.2:0.95~1.05;
2)控制温度-5℃~5℃,将羟基丙酰胺溶液、碱溶液以及硫酸二甲酯溶液通入微反应器中反应10-20分钟,得到R-2-氨基-N-苄基-3-甲氧基丙酰胺溶液;所述硫酸二甲酯、氢氧化钠与D-丝氨酸的摩尔比为2.00~2.05:4~4.5:1;
3)步骤2)得到的溶液流出微反应器,加入二氯甲烷萃取,然后将萃取后的二氯甲烷溶液(简称甲氧基丙酰胺溶液)与乙酸酐溶液通入微反应器中,控制温度在10℃~25℃,反应10~20分钟,得到拉科酰胺粗品溶液,出微反应器;所述乙酰化试剂与D-丝氨酸的摩尔比为0.9~1.0:1。
4)将上述粗品溶液经过水洗,干燥后减压蒸馏部分溶剂后,降温0℃~5℃析晶得到拉科酰胺纯品。
上述步骤(1)中的有机溶剂可以是乙腈、四氢呋喃,优选乙腈。
本发明的甲基化反应和乙酰化反应分为两个微反应器。
甲基化反应的微反应器包括三个控温单元(羟基丙酰胺溶液控温单元、氢氧化钠溶液控温单元、硫酸二甲酯溶液控温单元)、反应单元1,其中羟基丙酰胺溶液控温单元、氢氧化钠溶液控温单元、硫酸二甲酯溶液控温单元与均反应单元1相连。羟基丙酰胺溶液、氢氧化钠溶液和硫酸二甲酯溶液先分别进入控温单元的羟基丙酰胺溶液控温单元、氢氧化钠溶液控温单元、硫酸二甲酯溶液控温单元,控制温度在-5℃~5℃,然后进入反应单元1反应10-20分钟,反应液出微反应器后经过二氯甲烷萃取,得到甲氧基丙酰胺溶液。
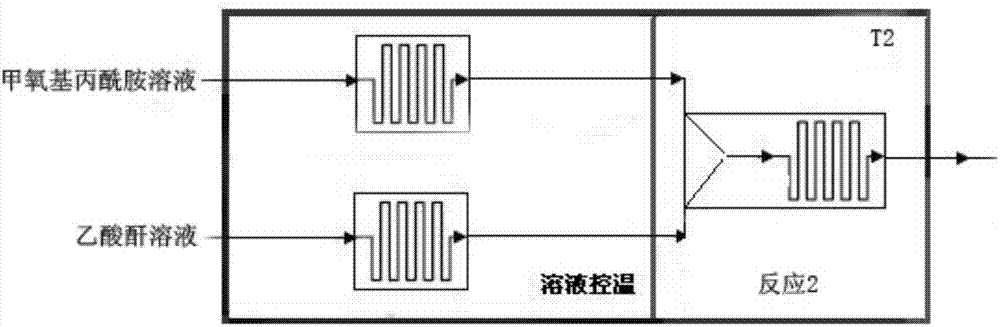
乙酰化反应的微反应器包括两个控温单元(甲氧基丙酰胺溶液控温单元、乙酸酐溶液控温单元)、反应单元2,其中甲氧基丙酰胺溶液控温单元、乙酸酐溶液控温单元均与反应单元2相连。甲氧基丙酰胺溶液与乙酸酐溶液先进入甲氧基丙酰胺二氯甲烷溶液控温单元、乙酸酐溶液控温单元控制温度10℃~25℃,然后进入反应单元2反应10-20分钟,出微反应器进行下一步操作。
本发明使用的微反应器为康宁(上海)管理有限公司生产的康宁低流量微通道反应器LFR。所述各溶液进入微反应器的流速控制在1-50ml/min。
所述乙酰化反应如果不采用微反应器,则采用通常的乙酰化步骤,具体为:步骤2)得到的溶液流出微反应器,加入二氯甲烷萃取,然后将萃取后的二氯甲烷溶液中滴加乙酸酐溶液,滴毕,控制温度在10℃~25℃反应1~3小时,得到拉科酰胺粗品溶液;所述乙酰化试剂与D-丝氨酸的摩尔比为0.9~1.0:1。
本发明的制备方法采用微反应器后,D-丝氨酸的氨基不再需要保护,其原理如下:本发明经过试验发现,该方法中缩合所用到的氯甲酸异丁酯能够选择性很好的与羧基结合生成混合酸酐,进而与苄胺发生缩合反应,而裸露的氨基氢在微反应器中的甲基化过程中不会与甲基化试剂反应,原因是羟基氢的化学活性比氨基氢要强,在微反应器中反应迅速,甲基化试剂优先与羟基氢反应,通过控制反应时间和控制甲基化试剂的用量,可以避免副产物的产生。但是在传统方法下,由于反应时间较长(5h左右)且甲基化试剂过量(一般过量3~4倍)就会导致副产物大量增加,D-丝氨酸必须预先进行氨基保护。
本发明的有益效果是:
1、相比传统方法,本发明D-丝氨酸的氨基不需要保护,合成路线由5步反应减缩为3步反应,大大简化了反应步骤。同时本发明采用微反应器进行反应,可实现每个反应单元的反应物料的充分混合和对反应的精确控制,大大减少了副产物的产生,实现了反应过程的连续性和自动化。相比传统的反应方式,本发明极大缩短了反应时间,由原来的9-10h减少至2-3h,且安全可控。
2、传统方法中,甲基化过程会出现局部放热现象,甚至会导致喷料,但是使用微反应器时,由于其具有有着极好的传热和传质能力,可以实现物料的瞬间均匀混合和高效的传热,不会出现上述喷料的现象。
3、本发明的析晶溶剂为后两步的反应溶剂,析晶方法简单,不需要引入其他溶剂;可回收循环利用,环境友好。
4、总之,采用本发明的方法,工艺步骤简单、反应条件温和、安全性好、环境友好,各步副产物少,产品收率高(总收率≥80%),且制备的拉科酰胺纯度达到99.9%以上,手性纯度达到99.95%,各步产品易于分离提纯,适合工业化生产。
附图说明
图1为本发明甲基化反应的微反应器的结构示意图;
图2为本发明乙酰化反应的微反应器的结构示意图。
具体实施方式
如图1所示。甲基化反应的微反应器包括三个控温单元(羟基丙酰胺溶液控温单元、氢氧化钠溶液控温单元、硫酸二甲酯溶液控温单元)、反应单元1,其中羟基丙酰胺溶液控温单元、氢氧化钠溶液控温单元、硫酸二甲酯溶液控温单元与均反应单元1相连。羟基丙酰胺溶液、氢氧化钠溶液、硫酸二甲酯溶液先分别进入控温单元的羟基丙酰胺溶液控温单元、氢氧化钠溶液控温单元、硫酸二甲酯溶液控温单元,控制温度在-5℃~5℃,然后进入反应单元1反应10-20分钟,反应液出微反应器后经过二氯甲烷萃取,得到甲氧基丙酰胺溶液。
如图2所示,乙酰化反应的微反应器包括两个控温单元(甲氧基丙酰胺溶液控温单元、乙酸酐溶液控温单元)、反应单元2,其中甲氧基丙酰胺溶液控温单元、乙酸酐溶液控温单元均与反应单元2相连。甲氧基丙酰胺溶液与乙酸酐溶液先进入甲氧基丙酰胺二氯甲烷溶液控温单元、乙酸酐溶液控温单元控制温度10℃~25℃,然后进入反应单元2反应10-20分钟,出微反应器进行下一步操作。
实施例1(R-2-氨基-N-苄基-3-羟基丙酰胺的合成)
将D-丝氨酸50g加入到100ml乙腈中,降温至-15℃,滴加52.9g三乙胺,滴毕搅拌10min,滴加65g氯甲酸异丁酯,滴毕搅拌10min,缓慢加入50.5g苄胺,滴毕,反应液直接用于下一步反应。
实施例2(R-2-氨基-N-苄基-3-甲氧基丙酰胺的合成)
实施例1的羟基丙酰胺溶液和30%的氢氧化钠溶液266g、120g硫酸二甲酯溶液先进入控温单元的羟基丙酰胺溶液控温单元、氢氧化钠溶液控温单元、硫酸二甲酯溶液控温单元,控制温度在-5℃~5℃,然后进入反应单元1反应10-20分钟,反应完后流出微反应器,经过二氯甲烷萃取,得到甲氧基丙酰胺的二氯甲烷溶液。
实施例3(R-2-乙酰氨基-N-苄基-3-甲氧基丙酰胺的合成)
实施例2的甲氧基丙酰胺二氯甲烷溶液与42g乙酸酐溶液先进入甲氧基丙酰胺二氯甲烷溶液控温单元、乙酸酐溶液控温单元,控温10℃~25℃,然后进入反应单元2反应10-20分钟,出微反应器进行下一步操作。
将上述反应液加入150ml水洗,二氯甲烷相用无水硫酸钠干燥、抽滤,将二氯甲烷减压蒸馏1/2得到拉科酰胺溶液。降温至0-5℃析晶0.5h,抽滤,30ml二氯甲烷淋洗滤饼。50-60℃鼓风烘干得产品97.38g。纯度99.97%,手性纯度99.96%,收率81.8%。
实施例4:(乙酰化反应不采用微反应器)
实施例2的甲氧基丙酰胺二氯甲烷溶液中滴加42g乙酸酐溶液,滴毕,控温10℃~25℃反应2h。
反应完成后,将上述反应液加入150ml水洗,二氯甲烷相用无水硫酸钠干燥、抽滤,将二氯甲烷减压蒸馏1/2得到拉科酰胺溶液。降温至0-5℃析晶0.5h,抽滤,30ml二氯甲烷淋洗滤饼。50-60℃鼓风烘干得产品95.22g。纯度99.90%,手性纯度99.92%,收率80.0%。
对比例(全部不采用微反应器):
将D-丝氨酸50g加入到200ml乙腈中,降温至-15℃,滴加52.9g三乙胺,滴毕搅拌10min,滴加65g氯甲酸异丁酯,滴毕搅拌10min,缓慢加入50.5g苄胺,滴毕,将滤液降温至-10℃-0℃,同时滴加氢氧化钠溶液与硫酸二甲酯溶液,滴毕反应7h,加入二氯甲烷萃取,取有机相。控制温度在10-25℃,向上述有机相中滴加乙酸酐42g,滴毕,反应2h,加入150ml水洗,二氯甲烷相用无水硫酸钠干燥、抽滤,将二氯甲烷减压蒸馏1/2得到拉科酰胺溶液。降温至0-5℃析晶0.5h,抽滤,30ml二氯甲烷淋洗滤饼。50-60℃鼓风烘干得产品55.45g。纯度99.81%,手性纯度99.80%,收率46.6%。
- 还没有人留言评论。精彩留言会获得点赞!