一种紫杉烷类化合物及其中间体的制备方法与流程
本发明属于药物化合物及其中间体合成技术领域,涉及一种紫杉烷类化合物及其中间体的制备方法,尤其涉及一种TPI-287及其中间体的制备方法。
背景技术:
紫杉烷类化合物是从植物中得到的一大类天然产物,而紫杉醇作为这类化合物的明星分子,最初也是从植物中提取后纯化得到的。随着紫杉烷类化合物的增多,对已发现的化合物进行结构改造,以提高生物活性、降低毒副作用,寻找疗效、耐受性更好、吸收更容易的类似物成为新的研究热点。
TPI-287,分子式为C46H63NO15,具有式A所示结构。TPI-287是继紫杉醇(Paclitaxel)、多西他赛(Docetaxel)、卡巴他赛(Cabazitaxel)后的第三代紫杉烷类抗癌药,可用于对多种药物产生耐药性的顽固性实体瘤的治疗,目前正处于临床研究阶段。
近三十年来,世界癌症发病率以年均3%~5%的速度递增,癌症已成为人类第一位死因,如何有效预防和治疗癌症一直是医学研究的重点课题。与紫杉醇、多西他赛等紫杉烷类抗癌药一样,TPI-287具有促进微管蛋白聚合,稳定微管并抑制其解聚的特性。体外细胞实验研究发现,TPI-287可以对乳腺癌、子宫癌及非小细胞肺癌等细胞系产生抑制作用,起到与其它紫杉烷类及大环内酯类抗癌药物类似的疗效。TPI-287在特定的动物肿瘤移植模型中表现出抑制肿瘤生长的特性,其优势在于,TPI-287有能力避开普通的抗药机制,具有更好的耐药性,还可渗透通过血-脑屏障,渗透中枢神经系统。TPI-287作为治疗多发性实体瘤耐药的换代产品,其市场前景广阔。
目前,关于TPI-287的合成方法报道较少,通常都是10-脱乙酰基巴卡亭III(又称10-DAB)作为起始原料,进行后续合成的,得到目标化合物TPI-287的,但往往存在中间体收率低且纯化困难,最终造成母环10-DAB的利用率低,成本高,不适用于工业化生产;或是其他反应物结构复杂,副反应多,影响收率,导致侧链使用量大,不利于节约成本等等问题。
因此,通过对TPI-287及其中间体化合物的合成方法进行研究,为TPI-287的合成提供不同的可能性,得到更适用于工业化生产的TPI-287及其中间体的合成方法具有重要的意义,也是领域内具有前瞻性的生产企业和一线研究人员亟待解决的问题。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种TPI-287及其中间体的制备方法,本发明提供的制备方法反应步骤较为简单,反应条件可控,反应原料廉价易得,而且具有较高的收率,适合规模化大生产。
本发明提供了一种TPI-287中间体A的制备方法,包括以下步骤:
1)在有机碱的作用下,将10-脱乙酰基巴卡亭III与羟基保护剂进行缩合反应,得到式(II)所示结构的化合物;
所述10-脱乙酰基巴卡亭III具有式(I)所示的结构;
其中,R为羟基保护基;
2)在氧化剂的作用下,将上述步骤得到的式(II)所示结构的化合物进行氧化反应,得到TPI-287中间体A;
所述TPI-287中间体A具有式(III)所示的结构;
其中,R为羟基保护基。
优选的,所述有机碱包括吡啶、三乙胺、N,N-二异丙基乙胺、吗啉和N-甲基吗啉中的一种或多种;
所述羟基保护剂包括氯甲酸三氯乙酯和/或硅醚类保护剂;
所述10-脱乙酰基巴卡亭III与所述羟基保护剂的摩尔比为1:(1~2);
所述10-脱乙酰基巴卡亭III与所述有机碱的摩尔比为1:(3~8);
所述缩合反应的温度为20~40℃;
所述缩合反应的时间为20~30h。
优选的,所述氧化反应包括Dess-Martin氧化、TPAP-NMO氧化、TEMPO氧化、Swern氧化、Oppenauer氧化、Moffatt-Pfitzner氧化、Sarett氧化或Corey-Kim氧化;
所述氧化剂包括Dess-Martin氧化剂、TPAP/NMO氧化剂、TEMPO、DMSO、(CH3)2CO、PCC氧化剂、无水氯化铜和醋酸铜中的一种或多种;
所述式(II)所示结构的化合物与所述氧化剂的摩尔比为1:(1.2~5);
所述氧化反应的温度为0~30℃;
所述氧化反应的时间为0.5~10h。
本发明提供了一种TPI-287中间体B的制备方法,包括以下步骤:
A)将上述技术方案任意一项所述的TPI-287中间体A和还原剂在有机溶剂中进行选择性还原反应,得到式(IV)所示结构的化合物;
其中,R为羟基保护基;
B)在有机碱的作用下,将上述步骤得到的式(IV)所示结构的化合物和乙酰化试剂经过乙酰化反应后,得到式(V)所示结构的化合物;
其中,R为羟基保护基;
C)在酸性条件下,将式(V)所示结构的化合物脱保护后,得到TPI-287中间体B;
所述TPI-287中间体B具有式(VI)所示的结构;
优选的,所述还原剂包括NaBH4、LiBH4和LiAlH4中的一种或多种;
所述TPI-287中间体A与所述还原剂的摩尔比为1:(2~3);
所述有机溶剂包括甲醇、乙醇和四氢呋喃中的一种或多种;
所述还原反应的温度为-10~30℃;
所述还原反应的时间为0.5~5h。
优选的,所述有机碱包括吡啶、三乙胺、N,N-二异丙基乙胺、吗啉和N-甲基吗啉中的一种或多种;
所述式(IV)所示结构的化合物与所述有机碱的摩尔比为1:(2~5);
所述乙酰化试剂包括乙酰氯、乙酸酐和冰乙酸中的一种或多种;
所述式(IV)所示结构的化合物与乙酰化试剂的摩尔比为1:(2~3);
所述乙酰化反应的温度为-20~25℃;
所述乙酰化反应的时间为10~30h。
优选的,所述酸性条件由HF-TEA、HF-Pyr、CF3COOH、PTSA和CSA中的一种或多种提供;
所述酸性条件为pH值小于等于4;
所述脱保护的时间为3~20h;
所述脱保护的温度为20~60℃。
本发明提供了一种TPI-287中间体C的制备方法,包括以下步骤:
将上述技术方案任意一项所述的TPI-287中间体B、式(VII)所示结构的化合物和有机酸进行反应后,得到TPI-287中间体C;
所述TPI-287中间体C具有式(VIII)所示的结构;
9、根据权利要求1所述的制备方法,其特征在于,所述有机酸包括CSA、PTSA和TFA中的一种或多种;
所述TPI-287中间体B、所述式(VII)所示结构的化合物与有机酸的摩尔比为1:(5~10):(0.05~0.1);
所述反应的时间为2~8h;
所述反应的温度为-20~30℃。
本发明还提供了一种TPI-287的制备方法,包括以下步骤:
将上述技术方案任意一项所述的TPI-287中间体C经过偶联和脱保护后,得到TPI-287。
本发明提供了一种TPI-287中间体A的制备方法,包括以下步骤,首先在有机碱的作用下,将10-脱乙酰基巴卡亭III与羟基保护剂进行缩合反应,得到式(II)所示结构的化合物;所述10-脱乙酰基巴卡亭III具有式(I)所示的结构;其中,R为羟基保护基;然后在氧化剂的作用下,将上述步骤得到的式(II)所示结构的化合物进行氧化反应,得到TPI-287中间体A;所述TPI-287中间体A具有式(III)所示的结构;其中,R为羟基保护基。与现有技术相比,本发明针对现有的TPI-287的合成过程中,存在中间体收率低且纯化困难,以及其他反应物结构复杂,副反应多,影响收率等问题,本发明从原料10-脱乙酰基巴卡亭III(又称10-DAB)的起始步骤入手,首先保护了10-DAB的C-7位羟基,然后进行选择性氧化反应,得到了TPI-287中间体A,为后续步骤提供了良好的基础,解决了中间体收率低且纯化困难等问题,本发明提供的制备方法反应步骤较为简单,反应条件可控,反应原料廉价易得,而且具有较高的收率,适合规模化大生产。本发明制备的TPI-287中间体A作为合成TPI-287的关键中间体,可进一步用于制备具有抗癌活性的潜在抗癌药物TPI-287。
实验结果表明,本发明提供的制备方法反应,从10-脱乙酰基巴卡亭III制备各个中间体到TPI-287,步骤简单,反应条件可控,反应原料廉价易得,适合规模化大生产,而且具有较高的收率和纯度。
附图说明
图1为本发明实施例1制备得到的化合物的HPLC谱图;
图2为本发明实施例4制备得到的化合物的HPLC谱图;
图3为本发明实施例7制备得到的化合物的HPLC谱图;
图4为本发明实施例9制备得到的化合物的HPLC谱图;
图5为本发明实施例11制备得到的化合物的HPLC谱图;
图6为本发明实施例13制备得到的化合物的HPLC谱图;
图7为本发明实施例4制备得到的化合物的质谱图。
具体实施方式
为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为了进一步说明本发明的特征和优点,而不是对发明权利要求的限制。
本发明所有原料和试剂,对其来源没有特别限制,在市场上购买的或按照本领域技术人员熟知的常规方法制备的即可。
本发明所有原料,对其纯度没有特别限制,本发明优选采用医药纯或紫杉烷类化合物制备领域的常规纯度。
本发明对所有结构式的表示方法没有特别限制,以本领域技术人员熟知的常规表示方法为基准,符合领域内的基本常识,本领域技术人员能够正确的理解其含义。
本发明提供了一种TPI-287中间体A的制备方法,包括以下步骤:
1)在有机碱的作用下,将10-脱乙酰基巴卡亭III与羟基保护剂进行缩合反应,得到式(II)所示结构的化合物;
所述10-脱乙酰基巴卡亭III具有式(I)所示的结构;
其中,R为羟基保护基;
2)在氧化剂的作用下,将上述步骤得到的式(II)所示结构的化合物进行氧化反应,得到TPI-287中间体A;
所述TPI-287中间体A具有式(III)所示的结构;
其中,R为羟基保护基。
本发明首先在有机碱的作用下,将10-脱乙酰基巴卡亭III与羟基保护剂进行缩合反应,得到式(II)所示结构的化合物;
所述10-脱乙酰基巴卡亭III具有式(I)所示的结构;
其中,R为羟基保护基;
本发明式(II)所示结构的化合物中的R为羟基保护基,本发明对其具体选择没有特别限制,以本领域技术人员熟知的常规羟基保护基即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述R来源于羟基保护剂,R的选择取决于羟基保护剂的选择,两者优选为对应关系。
本发明对所述10-脱乙酰基巴卡亭III的结构没有特别限制,以本领域技术人员熟知的10-脱乙酰基巴卡亭III的结构即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述10-脱乙酰基巴卡亭III优选具有式(I)所示的结构。
本发明对所述有机碱没有特别限制,以本领域技术人员熟知的此类反应的常用有机碱即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述有机碱优选包括吡啶、三乙胺、N,N-二异丙基乙胺、吗啉和N-甲基吗啉中的一种或多种,更优选为吡啶、三乙胺、N,N-二异丙基乙胺、吗啉或N-甲基吗啉。
本发明对所述有机碱的用量没有特别限制,以本领域技术人员熟知的此类反应的常规用量即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述10-脱乙酰基巴卡亭III与所述有机碱的摩尔比优选为1:(3~8),更优选为1:(4~7),最优选为1:(5~6)。
本发明对所述羟基保护剂没有特别限制,以本领域技术人员熟知的此类反应的常用羟基保护剂即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述羟基保护剂优选为能够在酸性条件下脱除保护基团的羟基保护剂,更优选包括氯甲酸三氯乙酯和/或硅醚类保护剂,最优选为氯甲酸三氯乙酯或硅醚类保护剂。本发明对所述硅醚类保护剂没有特别限制,以本领域技术人员熟知的此类反应的常用硅醚类保护剂即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述硅醚类保护剂优选包括三乙基氯硅烷、三甲基氯硅烷、三氟甲磺酸三甲基硅酯、叔丁基二苯基氯硅烷、叔丁基二甲基氯硅烷和三异丙基氯硅烷中的一种或多种;更优选为三乙基氯硅烷、三甲基氯硅烷、三氟甲磺酸三甲基硅酯、叔丁基二苯基氯硅烷、叔丁基二甲基氯硅烷或三异丙基氯硅烷,最优选为三乙基氯硅烷。
本发明对所述羟基保护剂的用量没有特别限制,以本领域技术人员熟知的此类反应的常规用量即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述10-脱乙酰基巴卡亭III与所述羟基保护剂的摩尔比优选为1:(1~2),更优选为1:(1.2~1.8),更优选为1:(1.3~1.7),最优选为1:(1.4~1.6)。
本发明对所述缩合反应的反应温度没有特别限制,以本领域技术人员熟知的此类反应的反应温度即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述缩合反应的温度优选为20~40℃,更优选为24~36℃,最优选为28~32℃。本发明对所述缩合反应的反应时间没有特别限制,以本领域技术人员熟知的此类反应的反应时间即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述缩合反应的时间优选为20~30h,更优选为22~28h,最优选为24~26h。
本发明对所述缩合反应的其他反应条件没有特别限制,以本领域技术人员熟知的此类反应的反应条件即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述缩合反应可以在保护性气体氛围或常态下进行,其中,所述保护性气体氛围优选包括氮气和/或惰性气体,更优选为氮气或氩气;所述常态优选为常压非隔绝空气状态。
本发明随后在氧化剂的作用下,将上述步骤得到的式(II)所示结构的化合物进行氧化反应,得到TPI-287中间体A。
本发明式(III)所示结构的化合物中的R为羟基保护基,本发明对其具体选择没有特别限制,以本领域技术人员熟知的常规羟基保护基即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述R来源于羟基保护剂,R的选择取决于羟基保护剂的选择,两者优选为对应关系。
本发明所述具有式(III)所示结构的TPI-287中间体A没有特别限制,以本领域技术人员熟知的制备TPI-287过程的某个中间体即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整。
本发明对所述氧化反应的类型没有特别限制,以本领域技术人员熟知的常规氧化类型即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述氧化反应的类型优选为TPI-287制备过程中的常见氧化反应类型,更优选为能将仲醇氧化为酮的氧化反应,更优选包括Dess-Martin氧化、TPAP-NMO氧化、TEMPO氧化、Swern氧化、Oppenauer氧化、Moffatt-Pfitzner氧化、Sarett氧化或Corey-Kim氧化,最优选为Dess-Martin氧化反应、TPAP-NMO氧化反应或TEMPO氧化反应。
本发明对所述氧化剂没有特别限制,以本领域技术人员熟知的此类反应的常用氧化剂即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述氧化剂优选为上述氧化反应类型的常用氧化剂,更优选包括Dess-Martin氧化剂、TPAP/NMO氧化剂、TEMPO、DMSO、(CH3)2CO、PCC氧化剂、无水氯化铜和醋酸铜中的一种或多种,更优选为Dess-Martin氧化剂、TPAP/NMO氧化剂、TEMPO、DMSO、(CH3)2CO、PCC氧化剂、无水氯化铜或醋酸铜,最优选为Dess-Martin氧化剂、TPAP/NMO(TPAP-NMO)氧化剂或TEMPO。
本发明对所述氧化剂的用量没有特别限制,以本领域技术人员熟知的此类反应的常规用量即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述式(II)所示结构的化合物与所述氧化剂的摩尔比优选为1:(1.2~5),更优选为1:(1.7~4.5),更优选为1:(2.2~4),最优选为1:(2.7~3.5)。
本发明对所述氧化反应的反应温度没有特别限制,以本领域技术人员熟知的此类反应的反应温度即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述氧化反应的温度优选为0~30℃,更优选为5~25℃,最优选为10~25℃。本发明对所述氧化反应的反应时间没有特别限制,以本领域技术人员熟知的此类反应的反应时间即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述氧化反应的时间优选为0.5~10h,更优选为2.5~8h,最优选为4.5~6h。
本发明提供了一种TPI-287中间体B的制备方法,包括以下步骤:
A)将上述技术方案任意一项所述的TPI-287中间体A和还原剂在有机溶剂中进行选择性还原反应,得到式(IV)所示结构的化合物;
其中,R为羟基保护基;
B)在有机碱的作用下,将上述步骤得到的式(IV)所示结构的化合物和乙酰化试剂经过乙酰化反应后,得到式(V)所示结构的化合物;
其中,R为羟基保护基;
C)在酸性条件下,将式(V)所示结构的化合物脱保护后,得到TPI-287中间体B;
所述TPI-287中间体B具有式(VI)所示的结构;
本发明首先将上述技术方案任意一项所述的TPI-287中间体A和还原剂在有机溶剂中进行选择性还原反应,得到式(IV)所示结构的化合物。
本发明式(IV)所示结构的化合物中的R为羟基保护基,本发明对其具体选择没有特别限制,以本领域技术人员熟知的常规羟基保护基即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述R来源于羟基保护剂,R的选择取决于羟基保护剂的选择,两者优选为对应关系。
本发明对所述还原剂没有特别限制,以本领域技术人员熟知的此类反应的常用还原剂即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述还原剂优选包括NaBH4、LiBH4和LiAlH4中的一种或多种,更优选为NaBH4、LiBH4或LiAlH4。
本发明对所述还原剂的用量没有特别限制,以本领域技术人员熟知的此类反应的常规用量即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述TPI-287中间体A与所述还原剂的摩尔比为1:(2~3),更优选为1:(2.2~2.8),最优选为1:(2.4~2.6)。
本发明对所述有机溶剂没有特别限制,以本领域技术人员熟知的此类反应的常用有机溶剂即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述有机溶剂优选包括甲醇、乙醇和四氢呋喃中的一种或多种,更优选为甲醇、乙醇或四氢呋喃。
本发明对所述有机溶剂的用量没有特别限制,以本领域技术人员熟知的此类反应的常规用量即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整。
本发明对所述选择性还原反应的反应温度没有特别限制,以本领域技术人员熟知的此类反应的反应温度即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述选择性还原反应的温度优选为-10~30℃,更优选为-5~25℃,更优选为0~20℃,最优选为5~15℃。本发明对所述选择性还原反应的反应时间没有特别限制,以本领域技术人员熟知的此类反应的反应时间即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述选择性还原反应的时间优选为0.5~5h,更优选为1.5~4h,最优选为2.5~3h。
本发明对所述选择性还原反应的其他反应条件没有特别限制,以本领域技术人员熟知的此类反应的反应条件即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述选择性还原反应可以在保护性气体氛围或常态下进行,其中,所述保护性气体氛围优选包括氮气和/或惰性气体,更优选为氮气或氩气;所述常态优选为常压非隔绝空气状态。
本发明随后在有机碱的作用下,将上述步骤得到的式(IV)所示结构的化合物和乙酰化试剂经过乙酰化反应后,得到式(V)所示结构的化合物。
本发明式(V)所示结构的化合物中的R为羟基保护基,本发明对其具体选择没有特别限制,以本领域技术人员熟知的常规羟基保护基即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述R来源于羟基保护剂,R的选择取决于羟基保护剂的选择,两者优选为对应关系。
本发明对所述有机碱没有特别限制,以本领域技术人员熟知的此类反应的常用有机碱即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述有机碱优选包括吡啶、三乙胺、N,N-二异丙基乙胺、吗啉和N-甲基吗啉中的一种或多种,更优选为吡啶、三乙胺、N,N-二异丙基乙胺、吗啉或N-甲基吗啉。
本发明对所述有机碱的用量没有特别限制,以本领域技术人员熟知的此类反应的常规用量即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述式(IV)所示结构的化合物与所述有机碱的摩尔比优选为1:(2~5),更优选为1:(2.5~4.5),最优选为1:(3~4)。
本发明对所述乙酰化试剂没有特别限制,以本领域技术人员熟知的此类反应的常用乙酰化试剂即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述乙酰化试剂优选包括乙酰氯、乙酸酐和冰乙酸中的一种或多种,更优选为乙酰氯、乙酸酐或冰乙酸。
本发明对所述乙酰化试剂的用量没有特别限制,以本领域技术人员熟知的此类反应的常规用量即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述式(IV)所示结构的化合物与乙酰化试剂的摩尔比优选为1:(2~3),更优选为1:(2.2~2.8),最优选为1:(2.4~2.6)。
本发明对所述乙酰化反应的反应温度没有特别限制,以本领域技术人员熟知的此类反应的反应温度即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述乙酰化反应的温度优选为-20~25℃,更优选为-10~15℃,最优选为0~5℃。本发明对所述乙酰化反应的反应时间没有特别限制,以本领域技术人员熟知的此类反应的反应时间即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述乙酰化反应的时间优选为10~30h,更优选为14~26h,最优选为18~22h。
本发明对所述反应的其他反应条件没有特别限制,以本领域技术人员熟知的此类反应的反应条件即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述乙酰化反应可以在保护性气体氛围或常态下进行,其中,所述保护性气体氛围优选包括氮气和/或惰性气体,更优选为氮气或氩气;所述常态优选为常压非隔绝空气状态。
本发明最后在酸性条件下,将式(V)所示结构的化合物脱保护后,得到TPI-287中间体B。
本发明所述具有式(VI)所示结构的TPI-287中间体B没有特别限制,以本领域技术人员熟知的制备TPI-287过程的某个中间体即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整。
本发明对产生所述酸性条件的原料没有特别限制,以本领域技术人员熟知的此类反应的常用产生酸性条件的原料即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述酸性条件优选由HF-TEA、HF-Pyr、CF3COOH、PTSA和CSA中的一种或多种提供,更优选由HF-TEA(氢氟酸三乙胺)、HF-Pyr(氢氟酸吡啶)、CF3COOH、PTSA(对甲苯磺酸)或CSA(樟脑磺酸)提供。
本发明对所述产生所述酸性条件的原料的用量没有特别限制,以本领域技术人员熟知的此类反应的常规用量即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述酸性条件优选为pH值小于等于4,更优选为小于等于3.5,最优选为小于等于3。
本发明对所述脱保护反应的反应温度没有特别限制,以本领域技术人员熟知的此类反应的反应温度即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述脱保护反应的温度优选为20~60℃,更优选为30~50℃,最优选为35~45℃。本发明对所述脱保护反应的时间没有特别限制,以本领域技术人员熟知的此类反应的反应时间即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述脱保护反应的时间优选为3~20h,更优选为5~18h,最优选为8~15h。
本发明对所述脱保护的其他反应条件没有特别限制,以本领域技术人员熟知的此类反应的反应条件即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述脱保护可以在保护性气体氛围或常态下进行,其中,所述保护性气体氛围优选包括氮气和/或惰性气体,更优选为氮气或氩气;所述常态优选为常压非隔绝空气状态。
本发明提供了一种TPI-287中间体C的制备方法,包括以下步骤:
将上述技术方案任意一项所述的TPI-287中间体B、式(VII)所示结构的化合物和有机酸进行反应后,得到TPI-287中间体C;
所述TPI-287中间体C具有式(VIII)所示的结构;
本发明所述具有式(VIII)所示结构的TPI-287中间体C没有特别限制,以本领域技术人员熟知的制备TPI-287过程的某个中间体即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整。
本发明对式(VII)所示结构的化合物没有特别限制,以本领域技术人员熟知的具有式(VII)所示结构的化合物即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述式(VII)所示结构的化合物优选为3,3-二甲氧基-1-丙烯,两者在本发明中具有等同的概念。
本发明对所述式(VII)所示结构的化合物,即3,3-二甲氧基-1-丙烯的用量没有特别限制,以本领域技术人员熟知的此类反应的常规用量即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述TPI-287中间体B与所述式(VII)所示结构的化合物的摩尔比为1:(5~10),更优选为1:(6~9),最优选为1:(7~8)。
本发明对有机酸没有特别限制,以本领域技术人员熟知的此类反应的常用有机酸即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述有机酸优选包括CSA、PTSA和TFA中的一种或多种,更优选为CSA、PTSA或TFA(三氟乙酸)。
本发明对所述有机酸的用量没有特别限制,以本领域技术人员熟知的此类反应的常规用量即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述TPI-287中间体B与有机酸的摩尔比优选为1:(0.05~0.1),更优选为1:(0.1~0.9),更优选为1:(0.3~0.7),最优选为1:(0.4~0.6)。
本发明对所述反应的温度没有特别限制,以本领域技术人员熟知的此类反应的反应温度即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述反应的温度优选为-20~30℃,更优选为-10~20℃,最优选为0~10℃。本发明对所述反应的时间没有特别限制,以本领域技术人员熟知的此类反应的反应时间即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述反应的时间优选为2~8h,更优选为3~7h,最优选为4~6h。
本发明还提供了一种TPI-287的制备方法,包括以下步骤:
将上述技术方案任意一项所述的TPI-287中间体C经过偶联和脱保护后,得到TPI-287。
本发明对所述TPI-287中间体C制备得到TPI-287的过程和参数没有特别限制,以本领域技术人员熟知的常规过程和参数即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述TPI-287中间体C经过处理后,得到TPI-287。本发明所述处理优选包括偶联和脱保护,但不限于上述步骤。
本发明对所述TPI-287及其结构没有特别限制,以本领域技术人员熟知的TPI-287及其结构即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整。
本发明提供了一种TPI-287中间体A、B、C和TPI-287的制备方法,本发明针对现有的TPI-287的合成过程中,存在中间体收率低且纯化困难,以及其他反应物结构复杂,副反应多,影响收率等问题,本发明从原料10-脱乙酰基巴卡亭III(又称10-DAB)的起始步骤入手,首先保护了10-DAB的C-7位羟基,然后进行选择性氧化、还原反应,再乙酰化C-10位羟基,脱保护,最后进行缩醛化,解决了现有制备过程中存在的中间体收率低且纯化困难等问题,本发明提供的制备方法反应选择性好,收率高,操作简单,反应条件可控,反应原料廉价易得,而且具有较高的收率,适合规模化大生产。本发明制备的TPI-287中间体A、B和C作为合成TPI-287的关键中间体,可进一步用于制备具有抗癌活性的潜在抗癌药物TPI-287。
实验结果表明,本发明提供的制备方法反应,从10-脱乙酰基巴卡亭III制备各个中间体到TPI-287,步骤简单,反应条件可控,反应原料廉价易得,适合规模化大生产,而且具有较高的收率和纯度。
为了进一步理解本发明,下面结合实施例对本发明提供的一种紫杉烷类化合物及其中间体的制备方法进行说明,但是应当理解,这些实施例是在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制,本发明的保护范围也不限于下述的实施例。
本发明中提供的TPI-287中间体的制备方法中,所用原料或试剂均可由市场购得。
实施例1
式(II-a)所示化合物的制备
将10.0g 10-脱乙酰巴卡亭III母环,二氯甲烷25mL与无水吡啶40mL,加入250mL圆底烧瓶中完全溶解。氮气保护,室温下缓慢滴加三乙基氯硅烷6.0mL,滴毕,室温搅拌。HPLC监测,直至反应完成。
室温搅拌约20h后,加入水使反应停止;然后用乙酸乙酯萃取3次,合并有机相,用500mL饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至无馏出物。加正己烷打浆搅拌1h,过滤,减压真空干燥,即得式(II-a)所示化合物7-三乙基硅烷基保护母环11.2g,白色固体。
本发明实施例1制备的产物经HPLC检测,参见图1,图1为本发明实施例1制备的产物的HPLC图。由图1可知,其纯度为94.7%,收率92.2%。
实施例2
式(II-b)所示化合物的制备
将10.0g 10-脱乙酰巴卡亭III母环,二氯甲烷30mL与三乙胺40mL,加入250mL圆底烧瓶中完全溶解。20℃下缓慢滴加三异丙基氯硅烷7.5mL,滴毕,20℃搅拌。HPLC监测,直至反应完成。
20℃搅拌约23h后,加入水使反应停止;然后用乙酸乙酯萃取3次,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至无馏出物。加正己烷打浆搅拌1h,过滤,减压真空干燥,即得式(II-b)所示化合物7-三异丙基硅烷基保护母环11.7g,白色固体。
本发明实施例2制备的产物经HPLC检测,纯度为95.2%,收率90.8%。
实施例3
式(II-c)所示化合物的制备
将10.0g 10-脱乙酰巴卡亭III母环,吡啶70mL,加入250mL圆底烧瓶中完全溶解。35℃下缓慢滴加氯甲酸三氯乙酯4.0mL,滴毕,35℃搅拌。HPLC监测,直至反应完成。
35℃搅拌约27h后,加入水使反应停止;然后用乙酸乙酯萃取3次,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩。加正己烷打浆搅拌1h,过滤,减压真空干燥,即得式(II-c)所示化合物7-三氯乙基保护母环11.1g,白色固体。
本发明实施例3制备的产物经HPLC检测,纯度为94.2%,收率89.7%。
实施例4
式(III-a)所示化合物的制备
将按照实施例1制备的式(II-a)所示化合物10.0g,二氯甲烷150ml,加入500ml三口瓶中,置于0℃冷冻槽,滴加戴斯-马丁(Dess-Martin)氧化剂(0.03mol)的二氯甲烷溶液10ml,滴加完毕,室温搅拌反应,HPLC监测反应。室温搅拌反应约1h后,将反应液转至分液漏斗,依次用饱和NaHCO3溶液,饱和NaHSO3溶液,饱和NaCl溶液洗涤,收集有机相。用无水硫酸钠干燥,过滤,滤液减压浓缩,真空干燥箱减压干燥,得式(III-a)所示化合物9.6g。
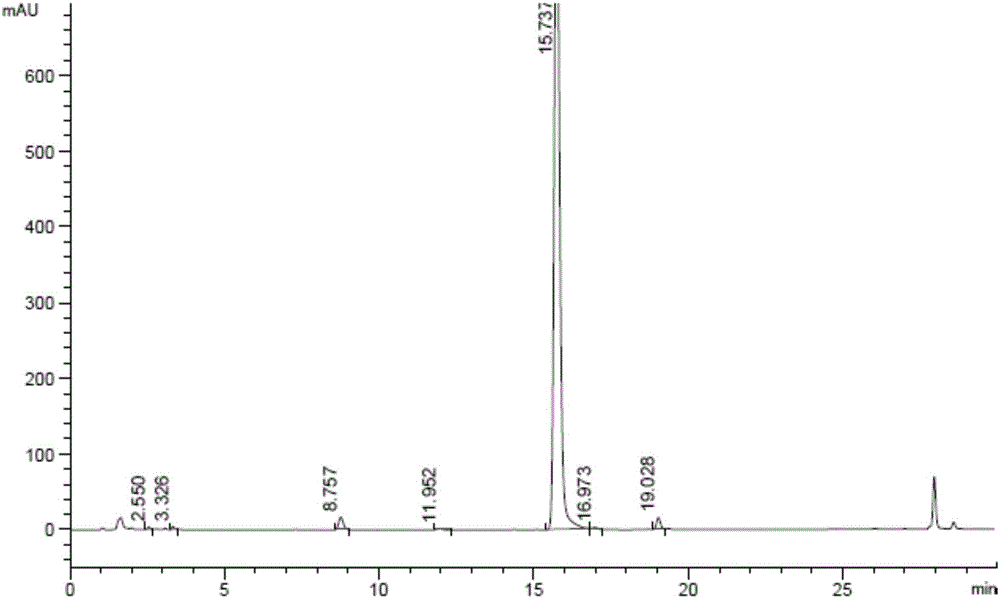
本发明实施例4制备的产物经HPLC检测,参见图2,图2为本发明实施例4制备的产物的HPLC图。由图2可知,经HPLC检测纯度为95.5%,收率96.3%。
参见图7,图7为本发明实施例4制备得到的化合物的质谱图。由图7可知,本发明制备得到了具有式(III-a)结构的化合物。
实施例5
式(III-b)所示化合物的制备
将按照实施例2制备的式(II-b)所示化合物10.0g,甲醇300ml,加入500ml圆底烧瓶中,充分搅拌溶解,30℃条件下,加入醋酸铜12.5g,搅拌反应10h,过滤,将滤液减压旋干,合并滤饼加入250ml水;然后用乙酸乙酯萃取3次,合并乙酸乙酯层浓缩至干,将蒸干品硅胶柱层析,得式(III-b)所示化合物6.5g。
本发明实施例5制备的产物经HPLC检测,经HPLC检测纯度为97.5%,收率66.3%。
实施例6
式(III-c)所示化合物的制备
将按照实施例3制备的式(II-c)所示化合物30.0g,少量溴化钾,二氯甲烷120ml,纯化水30ml,TEMPO氧化剂0.07g加入500ml反应瓶中,置于0℃冷冻槽,缓慢滴加7%NaHCO3水溶液120g,12.5%NaClO水溶液33g,剧烈搅拌约30min,转至分液漏斗萃取。有机层依次用包含0.76gNaI的10%NaHSO4溶液,10%Na2S2O3溶液及108.5g饱和食盐水洗涤,无水硫酸钠干燥,过滤,浓缩至无馏出物,经柱层析后得到式(III-c)所示化合物22.5g。
本发明实施例6制备的产物经HPLC检测,纯度为97.2%,收率75.2%。
实施例7
式(IV-a)所示化合物的制备
将按照实施例4制备的式(III-a)所示化合物4.0g,四氢呋喃20ml,甲醇4ml,加入250ml单口瓶中,完全溶解,将其置于-10℃冷冻槽中。氮气保护下,缓慢加入2.0M LiBH4的THF溶液7.2ml。滴加完毕后,室温搅拌反应1h,冰浴条件下,缓慢滴加10%醋酸铵的乙醇溶液15ml。然后加入0.1ml冰醋酸终止反应,向反应液中滴加纯水50ml,所得沉淀,经过滤,水洗,真空干燥,得到式(IV-a)所示化合物3.6g。
本发明实施例7制备的产物经HPLC检测,参见图3,图3为本发明实施例7制备的产物的HPLC图。由图3可知,经HPLC检测纯度为95.4%,收率91.1%。
实施例8
式(IV-c)所示化合物的制备
将按照实施例6制备的式(III-c)所示化合物10.0g,乙醇100ml,加入250ml单口瓶中,完全溶解,将其置于0℃冷冻槽中。氮气保护下,缓慢加入2.0M LiAlH4的THF溶液20.0ml。滴加完毕后,0~10℃搅拌反应4h,冰浴条件下,缓慢滴加10%醋酸铵的乙醇溶液40ml。然后加入冰醋酸终止反应,向反应液中滴加纯水,所得沉淀,经过滤,水洗,真空干燥,得到式(IV-c)所示化合物8.9g。
本发明实施例8制备的产物经HPLC检测,纯度为95.8%,收率89.2%。
实施例9
式(V-a)所示化合物的制备
将按照实施例7制备的式(IV-a)所示化合物3.6g,无水吡啶25ml,加入250ml单口瓶中,完全溶解,置于-20℃冷冻槽。氮气保护下,缓慢滴加乙酰氯的DMF溶液(1.0ml乙酰氯溶解于10mlDMF),滴加完毕,-20℃搅拌反应,HPLC监测,约20h反应完毕。将反应液倒入约5倍量的冰水中,搅拌,所得沉淀过滤,水洗,真空干燥,得到式(V-a)所示化合物3.34g。
本发明实施例9制备的产物经HPLC检测,参见图4,图4为本发明实施例9制备的产物的HPLC图。由图4可知,经HPLC检测,纯度为92.5%,收率87.4%。
实施例10
式(V-c)所示化合物的制备
将按照实施例8制备的式(IV-c)所示化合物5.0g,吗啉10ml,二氯甲烷50ml,加入250ml单口瓶中,完全溶解,置于0℃冷冻槽。氮气保护下,缓慢滴加乙酸酐的DMF溶液(2.0ml乙酸酐溶解于10mlDMF),滴加完毕,0~20℃搅拌反应,HPLC监测,约25h反应完毕。将反应液倒冰水中,二氯甲烷萃取2次,有机相用水洗,无水NaSO4干燥,减压浓缩,真空干燥,得到式(V-c)所示化合物4.4g。
本发明实施例10制备的产物经HPLC检测,纯度为94.1%,收率83.3%。
实施例11
式(VI)所示化合物的制备
将按照实施例9制备的式(V-a)所示化合物10.0g,80ml四氢呋喃加入250ml单口烧瓶中,搅拌溶解,室温下滴入氢氟酸三乙胺15ml,升温至50℃继续搅拌反应,HPLC监测,约3h反应完全。向反应液中加入适量冰水,滴加饱和NaOH溶液缓慢调节pH至7左右,然后加入固体NaHCO3至反应液呈碱性;加入乙酸乙酯萃取分液,收集有机相。然后用饱和NaCl溶液洗涤3次,无水硫酸钠干燥,过滤,滤液浓缩,真空干燥,得到式(VI)所示脱保护化合物8.1g。
本发明实施例11制备的产物经HPLC检测,参见图5,图5为本发明实施例11制备的产物的HPLC图。由图5可知,经HPLC检测,纯度为98.7%,收率96.8%。
实施例12
式(VI)所示化合物的制备
将按照实施例10制备的式(V-c)所示化合物10.0g,甲醇100ml,加入250ml单口烧瓶中,20~30℃搅拌溶解,加入对甲苯磺酸(PTSA),滴加水至反应液中,保证反应液无固体析出。滴毕,HPLC监测反应,反应15~20h后,加50mL乙酸乙酯及50mL水萃取,水层用乙酸乙酯洗涤,合并有机相。有机相用饱和食盐水洗涤,收集有机层减压旋干,经硅胶柱层析后,得到式(VI)所示化合物7.8g。
本发明实施例12制备的产物经HPLC检测,纯度为98.5%,收率94.1%。
实施例13
式(VIII)所示TPI-287中间体的制备
将式(VI)所示化合物10.0g,50ml二氯甲烷,加入250ml单口瓶中,室温搅拌溶清,向反应液中加入式(VII)所示化合物3,3-二甲氧基-1-丙烯12.1g,樟脑磺酸(CSA)0.175g,HPLC监测,3.5h后停止反应。加入125ml二氯甲烷稀释反应液,转移至盛有250ml 5%NaHCO3溶液的分液漏斗中,萃取,收集有机相,水相用二氯甲烷洗涤一次,合并有机层,用饱和氯化钠洗涤,无水Na2SO4干燥,抽滤,滤液减压浓缩,经硅胶柱层析,得到式(VIII)所示化合物9.1g。
本发明实施例13制备的产物经HPLC检测,参见图6,图6为本发明实施例13制备的产物的HPLC图。由图6可知,经HPLC检测,纯度为98.1%,收率85.1%。
由上述式(VIII)所示的中间体经过偶联和脱保护后,得到TPI-287。
实施例14
式(VIII)所示TPI-287中间体的制备
将式(VI)所示化合物10.0g,70ml二氯甲烷,加入250ml单口瓶中,-20℃搅拌溶清,向反应液中加入式(VII)所示化合物3,3-二甲氧基-1-丙烯15.6g,对甲苯磺酸(PTSA)0.3g,HPLC监测,7h后停止反应。加入100ml二氯甲烷稀释反应液,转移至盛有5%NaHCO3溶液的分液漏斗中,萃取,收集有机相,水相用二氯甲烷洗涤一次,合并有机层,用饱和氯化钠洗涤,无水Na2SO4干燥,抽滤,滤液减压浓缩,经硅胶柱层析,得到式(VIII)所示化合物8.4g。
本发明实施例14制备的产物经HPLC检测,纯度为98.4%,收率78.8%。
由上述式(VIII)所示的中间体经过偶联和脱保护后,得到TPI-287。
以上对本发明提供的TPI-287及其中间体的制备方法进行了详细的介绍,本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想,包括最佳方式,并且也使得本领域的任何技术人员都能够实践本发明,包括制造和使用任何装置或系统,和实施任何结合的方法。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。本发明专利保护的范围通过权利要求来限定,并可包括本领域技术人员能够想到的其他实施例。如果这些其他实施例具有不是不同于权利要求文字表述的结构要素,或者如果它们包括与权利要求的文字表述无实质差异的等同结构要素,那么这些其他实施例也应包含在权利要求的范围内。
- 还没有人留言评论。精彩留言会获得点赞!