光学活性1,5‑戊二醇衍生物及其合成方法和应用与流程
本发明属于合成医药化工领域,主要涉及光学活性1,5-戊二醇衍生物及其化学合成方法和应用。
背景技术:
1,5-戊二醇衍生物是重要的化工中间体,广泛用于聚酯、聚氨酯、增塑剂、涂料、香料、粘合剂、墨水、纤维、药物中间体、密封剂以及铅印等产品。近来,中国专利cn101316508a报道了1,5-戊二醇的组合可以用于减少和/或消除来自哺乳动物的气味,该气味如来自尿、粪便、腿部溃疡液体、褥疮性溃疡、血液和汗液的气味,可以用于制作吸收性产品或一次性卫生产品。因此合成1,5-戊二醇衍生物具有重要意义。
近年来已有大量文献报道了1,5-戊二醇衍生物的合成方法。中国专利cn1072168a报道了以取代的乙烯基乙醚和取代的丙烯醛反应,生成取代的3,4-二氢吡喃,取代的3,4-二氢吡喃酸催化水解生成取代的戊二醛,然后将镍负载在氧化铝上做催化剂进行加氢反应生成取代的戊二醇。中国专利cn101225022a报道了以镍-a/x负载型催化剂催化的戊二醛加氢反应获得戊二醇,其中组分a为co、mn、cu、cr等,载体x为分子筛、al2o3、sio2等。但是上述制取1,5-戊二醇衍生物的方法中,其共同存在的问题是催化加氢反应压力普遍都很高,这样势必对反应设备的压力要求较高,相应地就会使生成装置的一次性投资增高,生产成本增加,同时也会加大生产中的操作难度。同时,通过上述催化加氢法实现不对称的合成光学活性1,5-戊二醇衍生物也是很难的。不利于光学活性1,5-戊二醇衍生物在工业和有机合成中的应用及其工业化合成。
技术实现要素:
本发明克服了现有技术合成光学活性1,5-戊二醇衍生物的上述缺陷,提出了一种光学活性1,5-戊二醇衍生物及其制备方法。本发明制备方法中使用三组分的方法构建了比已有两组份方法更为复杂的光学活性1,5-戊二醇衍生物;本发明制备方法中使用的原料手性二芳基脯氨醇硅醚是天然存在的手性氨基酸衍生物,化学合成价廉易得;本发明制备方法具有高效原子经济性,高对映选择性,高产率,底物适用范围广,操作简单安全等有益效果。本发明制备的光学活性1,5-戊二醇衍生物具有高对映选择性,对hct116(人结肠癌细胞)有明显抑制作用,适用于抗肿瘤药物的制备应用。
本发明提出的光学活性1,5-戊二醇衍生物,其结构如式(ia)和式(ib)所示,
其中,ar1为芳基,选自于苯基、卤素取代的苯基、甲氧基取代的苯基、c1-c3烷基取代的苯基、硝基取代的苯基;
ar2为芳基,选自于苯基、卤素取代的苯基、甲氧基取代的苯基、c1-c3烷基取代的苯基;
ar3为芳基,选自于苯基、卤素取代的苯基、甲氧基取代的苯基、c1-c3烷基取代的苯基。
优选地,所述ar1为芳基,选自于苯基、4-氟苯基、4-氯苯基、4-溴苯基、3-溴苯基、4-甲氧基苯基、4-甲基苯基、4-硝基苯基、3-甲基苯基、2-甲基苯基、或3-甲氧基苯基;
ar2为芳基,选自于苯基、2-甲基苯基、2-溴苯基、3-甲基苯基、3-甲氧基苯基、4-甲基苯基、4-溴苯基、4-氟苯基、或4-甲氧基苯基;
ar3为芳基,选自于苯基、2-甲基苯基、2-溴苯基、3-甲基苯基、3-溴苯基、3-甲氧基苯基、4-甲基苯基、4-溴苯基、4-氟苯基、或4-甲氧基苯基。
进一步优选地,所述光学活性1,5-戊二醇衍生物选自:
(2s,3s)-2-苄氧基-2-苯基-3-间甲基苯基-1,5-戊二醇(1a)
(2r,3s)-2-苄氧基-2-苯基-3-间甲基苯基-1,5-戊二醇(1b)
(2s,3s)-2-苄氧基-2-苯基-3-对甲基苯基-1,5-戊二醇(2a)
(2r,3s)-2-苄氧基-2-苯基-3-对甲基苯基-1,5-戊二醇(2b)
(2s,3s)-2-苄氧基-2-苯基-3-对甲氧基苯基-1,5-戊二醇(3a)
(2r,3s)-2-苄氧基-2-苯基-3-对甲氧基苯基-1,5-戊二醇(3b)
(2s,3s)-2-对溴苄氧基-2,3-双苯基-1,5-戊二醇(4a)
(2r,3s)-2-对溴苄氧基-2,3-双苯基-1,5-戊二醇(4b)
(2s,3s)-2-间溴苄氧基-2,3-双苯基-1,5-戊二醇(5a)
(2r,3s)-2-间溴苄氧基-2,3-双苯基-1,5-戊二醇(5b)
(2s,3s)-2-对甲氧基苄氧基-2,3-双苯基-1,5-戊二醇(6a)
(2r,3s)-2-对甲氧基苄氧基-2,3-双苯基-1,5-戊二醇(6b)
(2s,3s)-2-苄氧基-2-间甲氧基苯基-3-苯基-1,5-戊二醇(7a)
(2r,3s)-2-苄氧基-2-间甲氧基苯基-3-苯基-1,5-戊二醇(7b)
(2s,3s)-2-苄氧基-2-邻溴苯基-3-苯基-1,5-戊二醇(8a)
(2r,3s)-2-苄氧基-2-邻溴苯基-3-苯基-1,5-戊二醇(8b)
(2s,3s)-2-苄氧基-2-对甲基苯基-3-苯基-1,5-戊二醇(9a)
(2r,3s)-2-苄氧基-2-对甲基苯基-3-苯基-1,5-戊二醇(9b)
(2s,3s)-2-苄氧基-2-对溴苯基-3-苯基-1,5-戊二醇(10a)
(2r,3s)-2-苄氧基-2-对溴苯基-3-苯基-1,5-戊二醇(10b)
本发明提出的光学活性1,5-戊二醇衍生物具有两个手性中心,包括式(ia)和式(ib)所示的一对光学活性1,5-戊二醇衍生物。
本发明还提出了所述式(ia)和式(ib)光学活性1,5-戊二醇衍生物的合成方法,在有机溶剂中,以式(1)α,β-不饱和醛、式(2)重氮化合物、式(3)苄醇衍生物为原料,以
具体地,所述方法包括以下步骤:在有机溶剂中,以式(1)α,β-不饱和醛、式(2)重氮化合物、式(3)苄醇衍生物为原料,以
其中,ar1为芳基,选自于苯基、卤素取代的苯基、甲氧基取代的苯基、c1-c3烷基取代的苯基、硝基取代的苯基;
ar2为芳基,选自于苯基、卤素取代的苯基、甲氧基取代的苯基、c1-c3烷基取代的苯基;
ar3为芳基,选自于苯基、卤素取代的苯基、甲氧基取代的苯基、c1-c3烷基取代的苯基。
优选地,所述ar1为芳基,选自于苯基、4-氟苯基、4-氯苯基、4-溴苯基、3-溴苯基、4-甲氧基苯基、4-甲基苯基、4-硝基苯基、3-甲基苯基、2-甲基苯基、或3-甲氧基苯基;
ar2为芳基,选自于苯基、2-甲基苯基、2-溴苯基、3-甲基苯基、3-甲氧基苯基、4-甲基苯基、4-溴苯基、4-氟苯基、或4-甲氧基苯基;
ar3为芳基,选自于苯基、2-甲基苯基、2-溴苯基、3-甲基苯基、3-溴苯基、3-甲氧基苯基、4-甲基苯基、4-溴苯基、4-氟苯基、或4-甲氧基苯基。
进一步优选地,所述光学活性1,5-戊二醇衍生物选自:
(2s,3s)-2-苄氧基-2-苯基-3-间甲基苯基-1,5-戊二醇(1a)
(2r,3s)-2-苄氧基-2-苯基-3-间甲基苯基-1,5-戊二醇(1b)
(2s,3s)-2-苄氧基-2-苯基-3-对甲基苯基-1,5-戊二醇(2a)
(2r,3s)-2-苄氧基-2-苯基-3-对甲基苯基-1,5-戊二醇(2b)
(2s,3s)-2-苄氧基-2-苯基-3-对甲氧基苯基-1,5-戊二醇(3a)
(2r,3s)-2-苄氧基-2-苯基-3-对甲氧基苯基-1,5-戊二醇(3b)
(2s,3s)-2-对溴苄氧基-2,3-双苯基-1,5-戊二醇(4a)
(2r,3s)-2-对溴苄氧基-2,3-双苯基-1,5-戊二醇(4b)
(2s,3s)-2-间溴苄氧基-2,3-双苯基-1,5-戊二醇(5a)
(2r,3s)-2-间溴苄氧基-2,3-双苯基-1,5-戊二醇(5b)
(2s,3s)-2-对甲氧基苄氧基-2,3-双苯基-1,5-戊二醇(6a)
(2r,3s)-2-对甲氧基苄氧基-2,3-双苯基-1,5-戊二醇(6b)
(2s,3s)-2-苄氧基-2-间甲氧基苯基-3-苯基-1,5-戊二醇(7a)
(2r,3s)-2-苄氧基-2-间甲氧基苯基-3-苯基-1,5-戊二醇(7b)
(2s,3s)-2-苄氧基-2-邻溴苯基-3-苯基-1,5-戊二醇(8a)
(2r,3s)-2-苄氧基-2-邻溴苯基-3-苯基-1,5-戊二醇(8b)
(2s,3s)-2-苄氧基-2-对甲基苯基-3-苯基-1,5-戊二醇(9a)
(2r,3s)-2-苄氧基-2-对甲基苯基-3-苯基-1,5-戊二醇(9b)
(2s,3s)-2-苄氧基-2-对溴苯基-3-苯基-1,5-戊二醇(10a)
(2r,3s)-2-苄氧基-2-对溴苯基-3-苯基-1,5-戊二醇(10b)
具体地,所述方法包括:(1)在有机溶剂中,以式(1)α,β-不饱和醛、式(2)重氮化合物、式(3)苄醇衍生物为原料,以
更具体地,所述方法包括:将α,β-不饱和醛、金属催化剂、手性二芳基脯氨醇硅醚、取代苯甲酸和
本发明方法中,所述合成光学活性1,5-戊二醇衍生物的反应的温度为0~50℃;优选地,为25℃、40℃。进一步优选地,所述反应中两步反应温度不同,步骤(1)反应的温度为0~40℃,步骤(2)反应的温度为20~50℃;优选地,步骤(1)反应的温度为25℃,步骤(2)反应的温度为40℃。
本发明方法中,式(2)重氮化合物:式(3)苄醇衍生物:式(1)α,β-不饱和醛:金属催化剂:手性二芳基脯氨醇硅醚:取代苯甲酸:金属负氢试剂的摩尔比为1.0-3.0:1.0-2.5:1.0:0.05-0.2:0.15-0.2:0.15-0.5:10。优选地,重氮化合物:苄醇衍生物:α,β-不饱和醛:金属催化剂:手性二芳基脯氨醇硅醚:取代苯甲酸:金属负氢试剂的摩尔比为3.0:2.5:1.0:0.2:0.2:0.5:10、1.0:1.0:1.0:0.05:0.15:0.15:10、2.0:2.0:1.0:0.1:0.2:0.4:10。
本发明方法中,还可以包括对反应得到的光学活性1,5-戊二醇衍生物进行分离纯化的步骤,第一步反应的分离纯化用体积比为乙酸乙酯:石油醚=1:500~1:80洗脱剂进行柱层析,第二步反应的分离纯化用体积比为乙酸乙酯:石油醚=1:50~1:8洗脱剂进行柱层析。优选地,第一步反应的分离纯化用体积比为乙酸乙酯:石油醚=1:100洗脱剂进行柱层析,第二步反应的分离纯化用体积比为乙酸乙酯:石油醚=1:10洗脱剂进行柱层析。
本发明方法中,所述有机溶剂包括二氯甲烷、四氢呋喃、甲苯或氯仿;优选地,为二氯甲烷。
本发明方法中,所述金属催化剂为铱复合物,铑复合物或钯复合物;优选地,所述金属催化剂为铱复合物;进一步优选地,为双(1,5-环辛二烯)四氟硼酸铱。
本发明方法中,所述手性二芳基脯氨醇硅醚结构如式(ⅲ)所示,
其中,r1为三甲基硅基,三乙基硅基或二甲基叔丁基硅基;ar4为苯基或3,5-双三氟甲基苯基。优选地,r1为二甲基叔丁基硅基,ar4为苯基。
本发明方法中,所述取代苯甲酸结构如式(ⅳ)所示,
其中,r包括氢、3,5-双三氟甲基、硝基、甲氧基、卤素。优选地,r为4-硝基。
本发明方法中,所述金属负氢试剂为四氢铝锂或硼氢化钠;优选地,为硼氢化钠。
在一个具体实施方案中,本发明中光学活性1,5-戊二醇衍生物的合成方法为:第一步反应,按重氮化合物、苄醇衍生物、α,β-不饱和醛、铱复合物、手性二芳基脯氨醇硅醚、取代苯甲酸摩尔比为2.0:2.0:1.0:0.1:0.2:0.4称取原料,以α,β-不饱和醛用量为基准量取
本发明中,所述如式(ia)和式(ib)所示的光学活性1,5-戊二醇衍生物为一对高对映选择性的化合物。
本发明还提出了由本发明所述合成方法制备得到的如式(ia)和式(ib)所示的光学活性1,5-戊二醇衍生物;所述式(ia)和式(ib)所示的光学活性1,5-戊二醇衍生物具有两个手性中心。
本发明还提出了如式(ia)和式(ib)所示的光学活性1,5-戊二醇衍生物;所述式(ia)和式(ib)所示的光学活性1,5-戊二醇衍生物具有两个手性中心。
本发明还提出了如式(ia)和式(ib)所示的光学活性1,5-戊二醇衍生物在制备医药中间体中的应用。
本发明还提出了如式(ia)和式(ib)所示的光学活性1,5-戊二醇衍生物在制备抗肿瘤药物中的应用。其中,所述肿瘤疾病包括结肠癌等。
本发明还提出了如式(ia)和式(ib)所示的一对高对映选择性的光学活性1,5-戊二醇衍生物在制备抗结肠癌药物中的应用。
本发明提出的具有两个手性中心的如式(ia)和式(ib)所示的一对光学活性1,5-戊二醇衍生物是重要的化工和医药中间体,在医药化工领域广泛应用,具有很大应用前景。本发明的制备方法以廉价易得的化合物为原料,具有反应条件温和、反应步骤少、反应快、成本低、产生的废物少、操作简单安全、原子经济性高、选择性高、产率高等有益效果。
附图说明
图1是实施例1所得产物(1a)的1hnmr示意图。
图2是实施例1所得产物(1a)的13cnmr示意图。
图3为实施例1所得产物(1a)的消旋产物液相图。
图4为实施例1所得产物(1a)的手性产物液相图。
图5是实施例1所得产物(1b)的1hnmr示意图。
图6是实施例1所得产物(1b)的13cnmr示意图。
图7为实施例1所得产物(1b)的消旋产物液相图。
图8为实施例1所得产物(1b)的手性产物液相图。
图9是实施例2所得产物(2a)的1hnmr示意图。
图10是实施例2所得产物(2a)的13cnmr示意图。
图11为实施例2所得产物(2a)的消旋产物液相图。
图12为实施例2所得产物(2a)的手性产物液相图。
图13是实施例2所得产物(2b)的1hnmr示意图。
图14是实施例2所得产物(2b)的13cnmr示意图。
图15为实施例2所得产物(2b)的消旋产物液相图。
图16为实施例1所得产物(2b)的手性产物液相图。
图17是实施例3所得产物(3a)的1hnmr示意图。
图18是实施例3所得产物(3a)的13cnmr示意图。
图19为实施例3所得产物(3a)的消旋产物液相图。
图20为实施例3所得产物(3a)的手性产物液相图。
图21是实施例3所得产物(3b)的1hnmr示意图。
图22是实施例3所得产物(3b)的13cnmr示意图。
图23为实施例3所得产物(3b)的消旋产物液相图。
图24为实施例3所得产物(3b)的手性产物液相图。
图25是实施例4所得产物(4a)的1hnmr示意图。
图26是实施例4所得产物(4a)的13cnmr示意图。
图27为实施例4所得产物(4a)的消旋产物液相图。
图28为实施例4所得产物(4a)的手性产物液相图。
图29是实施例4所得产物(4b)的1hnmr示意图。
图30是实施例4所得产物(4b)的13cnmr示意图。
图31为实施例4所得产物(4b)的消旋产物液相图。
图32为实施例4所得产物(4b)的手性产物液相图。
图33为实施例5所得产物(5a)的的1hnmr示意图。
图34为实施例5所得产物(5a)的13cnmr示意图。
图35为实施例5所得产物(5a)的消旋产物液相图。
图36为实施例5所得产物(5a)的手性产物液相图。
图37为实施例5所得产物(5b)的1hnmr示意图。
图38为实施例5所得产物(5b)的13cnmr示意图。
图39为实施例5所得产物(5b)的消旋产物液相图。
图40为实施例5所得产物(5b)的手性产物液相图。
图41为实施例6所得产物(6a)的1hnmr示意图。
图42为实施例6所得产物(6a)的13cnmr示意图。
图43为实施例6所得产物(6a)的消旋产物液相图。
图44为实施例6所得产物(6a)的手性产物液相图。
图45为实施例6所得产物(6b)的1hnmr示意图。
图46为实施例6所得产物(6b)的13cnmr示意图。
图47为实施例6所得产物(6b)的消旋产物液相图。
图48为实施例6所得产物(6b)的手性产物液相图。
图49为实施例7所得产物(7a)的1hnmr示意图。
图50为实施例7所得产物(7a)的13cnmr示意图。
图51为实施例7所得产物(7a)的消旋产物液相图。
图52为实施例7所得产物(7a)的手性产物液相图。
图53为实施例7所得产物(7b)的1hnmr示意图。
图54为实施例7所得产物(7b)的13cnmr示意图。
图55为实施例7所得产物(7b)的消旋产物液相图。
图56为实施例7所得产物(7b)的手性产物液相图。
图57为实施例8所得产物(8a)的1hnmr示意图。
图58为实施例8所得产物(8a)的13cnmr示意图。
图59为实施例8所得产物(8a)的消旋产物液相图。
图60为实施例8所得产物(8a)的手性产物液相图。
图61为实施例8所得产物(8b)的1hnmr示意图。
图62为实施例8所得产物(8b)的13cnmr示意图。
图63为实施例8所得产物(8b)的消旋产物液相图。
图64为实施例8所得产物(8b)的手性产物液相图。
图65为实施例8所得产物(9a)的1hnmr示意图。
图66为实施例8所得产物(9a)的13cnmr示意图。
图67为实施例8所得产物(9a)的消旋产物液相图。
图68为实施例8所得产物(9a)的手性产物液相图。
图69为实施例8所得产物(9b)的1hnmr示意图。
图70为实施例8所得产物(9b)的13cnmr示意图。
图71为实施例8所得产物(9b)的消旋产物液相图。
图72为实施例8所得产物(9b)的手性产物液相图。
图73为实施例8所得产物(10a)的1hnmr示意图。
图74为实施例8所得产物(10a)的13cnmr示意图。
图75为实施例8所得产物(10a)的消旋产物液相图。
图76为实施例8所得产物(10a)的手性产物液相图。
图77为实施例8所得产物(10b)的1hnmr示意图。
图78为实施例8所得产物(10b)的13cnmr示意图。
图79为实施例8所得产物(10b)的消旋产物液相图。
图80为实施例8所得产物(10b)的手性产物液相图。
图81是不同浓度的两种本发明化合物(fb1-3-s、fb1-3-x)对hct116(人结肠癌细胞)的浓度-存活率曲线图。
具体实施方式
结合以下具体实施例和附图,对本发明作进一步的详细说明,本发明的保护内容不局限于以下实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。实施本发明的过程、条件、试剂、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。
本发明的光学活性1,5-戊二醇衍生物的制备方法,第一步反应,将α,β-不饱和醛、金属催化剂、手性二芳基脯氨醇硅醚、取代苯甲酸和
其中,ar1为芳基,选自于苯基、卤素取代的苯基、甲氧基取代的苯基、c1-c3烷基取代的苯基、硝基取代的苯基;
ar2为芳基,选自于苯基、卤素取代的苯基、甲氧基取代的苯基、c1-c3烷基取代的苯基;
ar3为芳基,选自于苯基、卤素取代的苯基、甲氧基取代的苯基、c1-c3烷基取代的苯基。
实施例1
第一步反应,将3-甲基肉桂醛(1.0mmol)、双(1,5-环辛二烯)四氟硼酸铱(0.1mmol)、手性二苯基脯氨醇二甲基叔丁基硅醚(0.2mmol)、4-硝基苯甲酸(0.4mmol)和
光学活性产物(1a)的1hnmr示意图如图1所示,其13cnmr示意图如图2所示,其消旋产物液相图如图3所示,其手性产物液相图如图4所示。
1hnmr(400mhz,cdcl3)δ7.34(dt,j=14.8,7.2hz,4h),7.23(td,j=5.6,3.8hz,3h),7.19(s,1h),6.96(d,j=3.7hz,2h),6.87(d,j=8.0hz,2h),6.57(d,j=6.8hz,2h),4.37(s,2h),3.86(s,2h),3.46(dd,j=11.6,3.2hz,1h),3.34(ddd,j=20.6,12.5,6.5hz,2h),2.45(ddd,j=10.1,6.7,3.9hz,1h),2.21(s,3h),1.93(s,1h),1.70–1.57(m,1h).
13cnmr(100mhz,cdcl3)δ138.59,137.29,136.48,136.07,129.92,128.58,128.40,127.96,127.66,127.58,127.14,84.08,77.32,77.01,76.69,64.85,62.00,61.83,47.60,31.69,21.03.
hplc(手性ic柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=33.13分钟,tminor=8.15分钟.
光学活性产物(1b)的1hnmr示意图如图5所示,其13cnmr示意图如图6所示,其消旋产物液相图如图7所示,其手性产物液相图如图8所示。
1hnmr(400mhz,cdcl3)δ7.39–7.25(m,6h),7.20(dd,j=12.4,4.5hz,4h),7.06(d,j=7.7hz,2h),6.99(d,j=7.6hz,2h),4.51(q,j=12.1hz,2h),4.08–3.91(m,2h),3.33–3.22(m,1h),3.11(dd,j=15.6,9.2hz,1h),2.99(dd,j=11.4,2.7hz,1h),2.23(d,j=14.4hz,3h),2.05–1.93(m,1h),1.86(s,1h),1.55–1.48(m,2h).
13cnmr(100mhz,cdcl3)δ140.51,139.39,136.62,136.55,129.91,128.80,128.44,128.41,127.34,127.17,126.88,126.75,84.15,77.34,77.02,76.71,65.71,65.66,61.18,52.10,32.81,21.06.
hplc(手性ic柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=7.39分钟,tminor=12.91分钟.
实施例2
第一步反应,将4-甲基肉桂醛(1.0mmol)、双(1,5-环辛二烯)四氟硼酸铱(0.1mmol)、手性二苯基脯氨醇二甲基叔丁基硅醚(0.2mmol)、4-硝基苯甲酸(0.4mmol)和
光学活性产物(2a)的1hnmr示意图如图9所示,其13cnmr示意图如图10所示,其消旋产物液相图如图11所示,其手性产物液相图如图12所示。
1hnmr(400mhz,cdcl3).δ7.39–7.27(m,4h),7.27–7.16(m,4h),6.99–6.84(m,4h),6.44(s,2h),4.37(s,2h),3.86(s,2h),3.44(dd,j=11.6,3.2hz,1h),3.36(ddd,j=11.8,7.5,4.7hz,1h),3.28(dd,j=16.3,8.7hz,1h),2.52–2.38(m,1h),2.11(s,3h),1.98(d,j=24.6hz,1h),1.71–1.61(m,1h),1.38–1.36(m,1h).
13cnmr(100mhz,cdcl3)δ139.15,138.62,137.38,137.05,130.88,128.58,127.99,127.67,127.61,127.59,127.49,127.16,84.04,77.35,77.04,76.72,64.86,62.03,61.79,47.95,31.66,21.34.
hplc(手性ic柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=17.58分钟,tminor=6.54分钟。
光学活性产物(2b)的1hnmr示意图如图13所示,其13cnmr示意图如图14所示,其消旋产物液相图如图15所示,其手性产物液相图如图16所示。
1hnmr(400mhz,cdcl3)δ7.44–7.26(m,9h),7.25–7.23(m,1h),7.14(t,j=7.7hz,1h),7.04(t,j=8.1hz,3h),4.59(dd,j=27.1,12.0hz,2h),4.08(s,2h),3.36(ddd,j=11.1,6.9,4.5hz,1h),3.20(dd,j=15.8,9.4hz,1h),3.08(dd,j=11.6,3.3hz,1h),2.29(s,3h),2.13–2.01(m,1h),1.93(ddd,j=8.9,6.9,3.4hz,1h),1.36–1.34(m,1h),1.09–1.07(m,1h).
13cnmr(100mhz,cdcl3)δ140.37,139.62,139.34,137.43,131.02,128.39,127.87,127.73,127.37,127.19,126.99,126.91,126.83,84.16,77.33,77.21,77.01,76.69,65.67,65.59,61.23,52.29,32.75,21.43.
hplc(手性ic柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=7.46分钟,tminor=9.88分钟.
实施例3
第一步反应,将4-甲氧基肉桂醛(1.0mmol)、双(1,5-环辛二烯)四氟硼酸铱(0.1mmol)、手性二苯基脯氨醇二甲基叔丁基硅醚(0.2mmol)、4-硝基苯甲酸(0.4mmol)和
光学活性产物(3a)的1hnmr示意图如图17所示,其13cnmr示意图如图18所示,其消旋产物液相图如图19所示,其手性产物液相图如图20所示。
1hnmr(400mhz,cdcl3)δ7.47–7.35(m,4h),7.29(t,j=14.9hz,4h),7.02(s,2h),6.67(s,4h),4.43(s,2h),3.93(s,2h),3.76(s,3h),3.52(dd,j=11.5,2.6hz,1h),3.44(dd,j=13.0,8.8hz,1h),3.40–3.29(m,1h),2.63–2.45(m,1h),2.07(s,1h),1.67(dd,j=15.0,9.0hz,2h).
13cnmr(100mhz,cdcl3)δ158.47,138.59,137.27,131.15,131.01,128.59,127.92,127.69,127.67,127.59,127.15,113.04,84.16,77.35,77.03,76.71,64.88,61.94,61.76,55.14,47.06,31.69.
hplc(手性ic柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=22.97分钟,tminor=8.82分钟.
光学活性产物(3b)的1hnmr示意图如图21所示,其13cnmr示意图如图22所示,其消旋产物液相图如图23所示,其手性产物液相图如图24所示。
1hnmr(400mhz,cdcl3)δ7.33(ddd,j=19.0,13.4,7.1hz,8h),7.24(d,j=7.6hz,2h),7.13(d,j=8.3hz,2h),6.79(d,j=8.4hz,2h),4.57(q,j=12.1hz,2h),4.07(s,2h),3.79(s,3h),3.46–3.29(m,1h),3.27–3.13(m,1h),3.08(dd,j=11.4,2.9hz,1h),1.99(dd,j=20.5,14.8hz,2h),1.66(s,2h).
13cnmr(100mhz,cdcl3)δ158.59,140.34,139.36,131.57,131.00,128.42,127.36,127.19,126.90,126.72,113.41,84.22,77.36,77.04,76.72,65.58,65.47,61.13,55.20,51.38,32.83.
hplc(手性ic柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=7.97分钟,tminor=12.59分钟.
实施例4
第一步反应,将肉桂醛(1.0mmol)、双(1,5-环辛二烯)四氟硼酸铱(0.1mmol)、手性二苯基脯氨醇二甲基叔丁基硅醚(0.2mmol)、4-硝基苯甲酸(0.4mmol)和
光学活性产物(4a)的1hnmr示意图如图25所示,其13cnmr示意图如图26所示,其消旋产物液相图如图27所示,其手性产物液相图如图28所示。
1hnmr(400mhz,cdcl3)δ7.50(d,j=7.7hz,2h),7.32(d,j=22.0hz,5h),7.21–7.07(m,3h),6.99(s,2h),6.76(s,2h),4.39(dd,j=25.7,11.8hz,2h),3.95(s,2h),3.55(d,j=10.6hz,1h),3.43(s,1h),3.33(d,j=6.6hz,1h),2.56–2.40(m,1h),2.12(s,1h),1.73(dd,j=18.4,13.0hz,2h).
13cnmr(100mhz,cdcl3)δ139.10,137.62,137.23,131.65,130.07,128.77,128.54,127.99,127.79,127.71,126.99,126.07,121.36,84.09,77.35,77.04,76.72,64.22,62.22,61.53,48.03,31.59.
hplc(手性ia柱,波长等于220纳米,正己烷:异丙醇=10:1,流速=1.0毫升/分钟),tmajor=26.81分钟,tminor=13.34分钟.
光学活性产物(4b)的1hnmr示意图如图29所示,其13cnmr示意图如图30所示,其消旋产物液相图如图31所示,其手性产物液相图如图32所示。
1hnmr(400mhz,cdcl3)δ7.50(d,j=7.9hz,2h),7.43–7.14(m,12h),4.53(dd,j=33.1,12.3hz,2h),4.06(q,j=12.5hz,2h),3.35(d,j=3.7hz,1h),3.25–3.07(m,2h),2.00(dd,j=23.2,17.4hz,2h),1.64(s,2h).
13cnmr(100mhz,cdcl3)δ139.94,139.59,138.33,131.49,130.08,128.52,128.36,128.02,127.51,127.07,126.82,120.94,84.33,77.35,77.03,76.71,65.58,65.03,60.97,51.97,32.71.
hplc(手性ia柱,波长等于220纳米,正己烷:异丙醇=8.0:1,流速=1.0毫升/分钟),tmajor=19.29分钟,tminor=14.99分钟.
实施例5
第一步反应,将肉桂醛(1.0mmol)、双(1,5-环辛二烯)四氟硼酸铱(0.1mmol)、手性二苯基脯氨醇二甲基叔丁基硅醚(0.2mmol)、4-硝基苯甲酸(0.4mmol)和
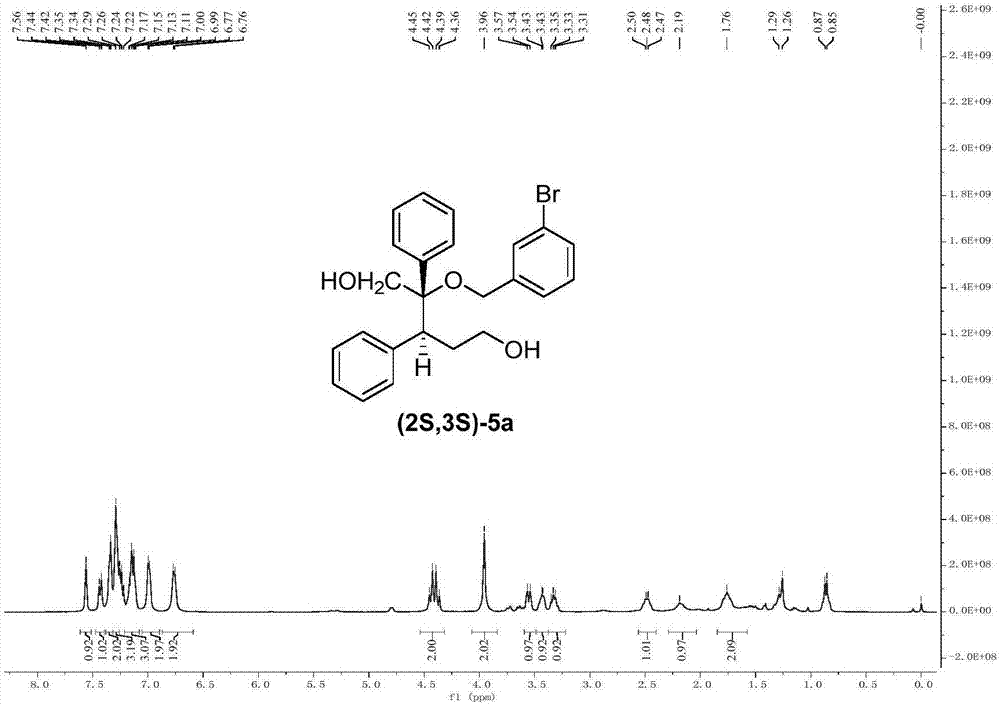
光学活性产物(5a)的1hnmr示意图如图33所示,其13cnmr示意图如图34所示,其消旋产物液相图如图35所示,其手性产物液相图如图36所示。
1hnmr(400mhz,cdcl3)δ7.56(s,1h),7.43(d,j=7.7hz,1h),7.34(d,j=5.0hz,2h),7.29(s,3h),7.14(dd,j=15.2,7.5hz,3h),6.99(d,j=4.6hz,2h),6.76(d,j=5.7hz,2h),4.41(dd,j=24.6,11.9hz,2h),3.96(s,2h),3.55(d,j=10.3hz,1h),3.43(d,j=3.6hz,1h),3.38–3.22(m,1h),2.56–2.40(m,1h),2.19(s,1h),1.76(s,2h).
13cnmr(100mhz,cdcl3)δ140.99,139.08,137.29,130.59,130.12,130.10,128.53,127.82,127.77,127.71,126.99,126.08,125.61,122.61,84.10,77.37,77.05,76.73,64.15,62.29,61.47,48.09,31.57.
hplc(手性ic柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1毫升/分钟),tmajor=6.15分钟,tminor=9.43分钟.
光学活性产物(5b)的1hnmr示意图如图37所示,其13cnmr示意图如图38所示,其消旋产物液相图如图39所示,其手性产物液相图如图40所示。
1hnmr(400mhz,cdcl3)δ7.54(s,1h),7.42(d,j=7.7hz,1h),7.36(t,j=7.3hz,2h),7.29(dd,j=14.8,7.2hz,5h),7.22(t,j=5.5hz,5h),4.55(dd,j=33.2,12.5hz,2h),4.15–3.99(m,2h),3.37(dd,j=9.5,5.2hz,1h),3.25–3.11(m,2h),2.03(dt,j=10.9,7.0hz,1h),1.95(dd,j=13.4,5.3hz,1h),1.62(s,1h),1.14(d,j=8.2hz,1h).
13cnmr(100mhz,cdcl3)δ141.67,139.76,139.46,130.22,130.12,129.93,129.68,128.53,128.06,127.53,127.12,126.83,125.12,122.58,84.37,77.34,77.02,76.70,65.50,64.89,60.99,51.80,32.67.
hplc(手性ia柱,波长等于220纳米,正己烷:异丙醇=20:1,流速=1.0毫升/分钟),tmajor=5.76分钟,tminor=9.71分钟.
实施例6
第一步反应,将肉桂醛(1.0mmol)、双(1,5-环辛二烯)四氟硼酸铱(0.1mmol)、手性二苯基脯氨醇二甲基叔丁基硅醚(0.2mmol)、4-硝基苯甲酸(0.4mmol)和
光学活性产物(6a)的1hnmr示意图如图41所示,其13cnmr示意图如图42所示,其消旋产物液相图如图43所示,其手性产物液相图如图44所示。
1hnmr(400mhz,cdcl3)δ7.36–7.28(m,5h),7.20–7.08(m,3h),7.02(d,j=4.2hz,2h),6.93(d,j=8.6hz,2h),6.74(d,j=5.7hz,2h),4.44–4.28(m,2h),3.92(s,2h),3.83(s,3h),3.55(dd,j=11.3,3.4hz,1h),3.43(ddd,j=11.7,7.4,4.7hz,1h),3.36(dd,j=15.9,9.1hz,1h),2.59–2.46(m,1h),2.07(s,1h),1.78–1.67(m,1h).
13cnmr(100mhz,cdcl3)δ159.20,139.38,137.29,130.56,130.07,128.81,127.98,127.67,127.31,126.90,114.04,84.00,77.34,77.02,76.70,64.64,61.94,61.80,55.32,48.02,31.71.
hplc(手性ic柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=38.04分钟,tminor=20.48分钟.
光学活性产物(6b)的1hnmr示意图如图45所示,其13cnmr示意图如图46所示,其消旋产物液相图如图47所示,其手性产物液相图如图48所示。
1hnmr(400mhz,cdcl3)δ7.47–7.28(m,6h),7.27–7.13(m,6h),6.93(d,j=8.5hz,2h),4.50(q,j=11.5hz,2h),4.19–3.98(m,2h),3.83(s,3h),3.45–3.28(m,1h),3.24–3.14(m,1h),3.11(dd,j=11.4,3.3hz,1h),2.13–1.86(m,2h).
13cnmr(100mhz,cdcl3)δ158.91,140.35,139.82,131.36,130.10,128.40,128.30,127.96,127.38,126.94,113.88,84.13,77.34,77.02,76.71,65.42,61.15,55.32,52.31,32.81.
hplc(手性ic柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=22.18分钟,tminor=38.56分钟.
实施例7
第一步反应,将肉桂醛(1.0mmol)、双(1,5-环辛二烯)四氟硼酸铱(0.1mmol)、手性二苯基脯氨醇二甲基叔丁基硅醚(0.2mmol)、4-硝基苯甲酸(0.4mmol)和
光学活性产物(7a)的1hnmr示意图如图49所示,其13cnmr示意图如图50所示,其消旋产物液相图如图51所示,其手性产物液相图如图52所示。
1hnmr(400mhz,cdcl3)δ7.46–7.27(m,4h),7.26–7.19(m,1h),7.16–6.96(m,4h),6.81–6.62(m,3h),6.58–6.39(m,2h),4.38(s,2h),3.93–3.76(m,2h),3.59(s,3h),3.48(dd,j=11.6,3.1hz,1h),3.41–3.30(m,1h),3.30–3.17(m,1h),2.52–2.35(m,1h),2.11(brs,1h),1.79–1.65(m,1h),1.53(brs,1h).
13cnmr(100mhz,cdcl3)δ158.05,138.23,138.07,137.60,129.12,127.63,127.54,126.60,126.53,126.07,125.89,119.23,112.67,112.16,83.02,63.94,61.17,60.62,54.10,47.01,30.56.
hplc(手性ia柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=9.22分钟,tminor=8.06分钟.
光学活性产物(7b)的1hnmr示意图如图53所示,其13cnmr示意图如图54所示,其消旋产物液相图如图55所示,其手性产物液相图如图56所示。
1hnmr(400mhz,cdcl3)δ7.40–7.34(m,4h),7.31–7.19(m,7h),6.86–6.79(m,2h),6.77(s,1h),4.58(q,j=12.2hz,2h),4.04(q,j=12.7hz,2h),3.64(s,3h),3.40–3.27(m,1h),3.24–3.08(m,2h),2.13–1.98(m,1h),1.99–1.86(m,1h),1.79(brs,1h),1.42(brs,1h).
13cnmr(100mhz,cdcl3)δ159.67,141.84,139.67,139.36,130.21,129.49,128.41,127.93,127.16,126.97,126.65,118.98,112.99,112.86,84.34,65.70,65.43,61.06,55.09,51.81,32.74.
hplc(手性ic柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=6.61分钟,tminor=10.59分钟.
实施例8
第一步反应,将肉桂醛(1.0mmol)、双(1,5-环辛二烯)四氟硼酸铱(0.1mmol)、手性二苯基脯氨醇二甲基叔丁基硅醚(0.2mmol)、4-硝基苯甲酸(0.4mmol)和
光学活性产物(8a)的1hnmr示意图如图57所示,其13cnmr示意图如图58所示,其消旋产物液相图如图59所示,其手性产物液相图如图60所示。
1hnmr(400mhz,cdcl3)δ7.54–7.36(m,4h),7.34–7.27(m,3h),7.22–7.08(m,3h),7.06–6.97(m,2h),6.88–6.60(m,2h),4.56–4.29(m,2h),3.94(d,j=4.8hz,2h),3.57(dd,j=11.6,3.2hz,1h),3.49–3.26(m,2h),2.63–2.46(m,1h),2.15–2.00(m,1h),1.82–1.67(m,1h),1.36(s,1h).
13cnmr(100mhz,cdcl3)δ139.27,138.57,137.27,130.10,128.59,127.92,127.71,127.67,127.60,127.15,126.93,84.04,64.88,62.05,61.73,48.02,31.66.
hplc(手性ia柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=15.82分钟,tminor=5.99分钟.
光学活性产物(8b)的1hnmr示意图如图61所示,其13cnmr示意图如图62所示,其消旋产物液相图如图63所示,其手性产物液相图如图64所示。
1hnmr(400mhz,cdcl3)δ7.45–7.35(m,5h),7.34–7.26(m,4h),7.25–7.18(m,5h),4.59(q,j=12.2hz,2h),4.17–3.98(m,2h),3.44–3.31(m,1h),3.25–3.16(m,1h),3.13(dd,j=11.6,3.3hz,1h),2.14–2.03(m,1h),2.01–1.90(m,1h),1.09(brs,1h).
13cnmr(100mhz,cdcl3)δ140.23,139.77,139.32,130.10,128.46,128.43,128.01,127.42,127.20,126.99,126.90,126.72,84.21,65.68,65.55,61.13,52.30,32.81.
hplc(手性ic柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=5.77分钟,tminor=8.11分钟.
实施例9
第一步反应,将肉桂醛(1.0mmol)、双(1,5-环辛二烯)四氟硼酸铱(0.1mmol)、手性二苯基脯氨醇二甲基叔丁基硅醚(0.2mmol)、4-硝基苯甲酸(0.4mmol)和
光学活性产物(9a)的1hnmr示意图如图65所示,其13cnmr示意图如图66所示,其消旋产物液相图如图67所示,其手性产物液相图如图68所示。
1hnmr(400mhz,cdcl3)δ7.53–7.32(m,4h),7.32–7.25(m,1h),7.21–7.01(m,5h),6.88(d,j=7.4hz,2h),6.83–6.52(m,2h),4.40(s,2h),3.88(s,2h),3.54(dd,j=11.3,2.5hz,1h),3.48–3.35(m,1h),3.35–3.21(m,1h),2.64–2.44(m,1h),2.34(s,3h),2.31–2.10(m,1h),1.91–1.49(m,2h).
13cnmr(100mhz,cdcl3)δ139.48,138.70,137.35,134.18,130.16,128.58,128.43,127.88,127.66,127.55,127.18,126.86,83.94,64.74,62.07,61.66,47.95,31.75,21.12.
hplc(手性ia柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=18.20分钟,tminor=6.11分钟.
光学活性产物(9b)的1hnmr示意图如图69所示,其13cnmr示意图如图70所示,其消旋产物液相图如图71所示,其手性产物液相图如图72所示。
1hnmr(400mhz,cdcl3)δ7.47–7.32(m,4h),7.30–7.26(m,1h),7.25–7.17(m,5h),7.15–7.01(m,4h),4.56(d,j=12.2hz,1h),4.50(d,j=12.2hz,1h),4.02(s,2h),3.46–3.26(m,1h),3.12(d,j=7.8hz,2h),2.32(s,3h),2.10–1.84(m,3h),1.69(brs,1h).
13cnmr(100mhz,cdcl3)δ139.81,139.48,137.03,136.88,130.28,129.10,128.41,127.88,127.14,126.96,126.88,126.69,84.13,65.46,65.26,60.96,51.71,32.77,21.08.
hplc(手性ia柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=5.91分钟,tminor=8.33分钟.
实施例10
第一步反应,将肉桂醛(1.0mmol)、双(1,5-环辛二烯)四氟硼酸铱(0.1mmol)、手性二苯基脯氨醇二甲基叔丁基硅醚(0.2mmol)、4-硝基苯甲酸(0.4mmol)和
光学活性产物(10a)的1hnmr示意图如图73所示,其13cnmr示意图如图74所示,其消旋产物液相图如图75所示,其手性产物液相图如图76所示。
1hnmr(400mhz,cdcl3)δ7.55–7.36(m,6h),7.35–7.30(m,1h),7.22–7.07(m,3h),6.89(d,j=8.2hz,2h),6.83–6.62(m,2h),4.42(dd,j=27.1,11.7hz,2h),3.90(s,2h),3.56(dd,j=11.7,3.0hz,1h),3.51–3.39(m,1h),3.39–3.25(m,1h),2.58–2.40(m,1h),2.11(brs,1h),1.78–1.65(m,1h),1.42(brs,1h).
13cnmr(100mhz,cdcl3)δ138.88,138.29,136.69,130.84,130.05,129.67,128.65,127.85,127.71,127.10,121.89,83.83,64.90,62.07,61.46,47.69,31.48.
hplc(手性ia柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=9.38分钟,tminor=6.88分钟.
光学活性产物(10b)的1hnmr示意图如图77所示,其13cnmr示意图如图78所示,其消旋产物液相图如图79所示,其手性产物液相图如图80所示。
1hnmr(400mhz,cdcl3)δ7.52–7.43(m,2h),7.42–7.34(m,4h),7.34–7.28(m,1h),7.27–7.19(m,5h),7.13(d,j=8.5hz,2h),4.54(dd,j=43.5,12.1hz,2h),4.05(s,2h),3.38(ddd,j=10.9,6.9,4.3hz,1h),3.27–3.16(m,1h),3.13(dd,j=11.7,3.3hz,1h),2.13–2.00(m,1h),1.98–1.86(m,1h),1.61(s,2h).
13cnmr(100mhz,cdcl3)δ139.50,139.39,138.97,131.50,130.09,128.79,128.49,128.10,127.34,127.13,126.67,121.50,83.96,65.65,65.20,60.94,51.84,32.62.
hplc(手性ic柱,波长等于220纳米,正己烷:异丙醇=4.5:1,流速=1.0毫升/分钟),tmajor=5.37分钟,tminor=4.66分钟。
实施例11
本实施例中用cck8法对本发明化合物生物活性进行测试,采用实施例10制备得到的本发明化合物(10a),(2s,3s)-2-苄氧基-2-对溴苯基-3-苯基-1,5-戊二醇(编号为fb1-3-s)和实施例10制备得到本发明化合物(10b),(2r,3s)-2-苄氧基-2-对溴苯基-3-苯基-1,5-戊二醇(编号为fb1-3-x)进行其对肿瘤细胞的抑制作用研究。本实施例具体使用的细胞系为:hct116(人结肠癌细胞)。
1、接种细胞:用含10%胎牛血清、1%青霉素和链霉素的mccoy'5a培养液配成单个细胞悬液,以每孔3000个细胞接种到96孔细胞培养板,每孔体积100μl。
2、经过碾磨处理步骤将本发明化合物制备为粉状药物,把粉状药物配制成终浓度为10mm的母液,而后梯度依次稀释至0.01mm,0.03mm,0.1mm,0.3mm,1mm,3mm,10mm,30mm。作用于细胞,每组浓度设置三个复孔。
3、给药:用完全培养基将梯度母液稀释至终浓度0.01μm,0.03μm,0.1μm,0.3μm,1μm,3μm,10μm,30μm,每孔200μl作用于细胞,每组浓度设置三个复孔,培养液中dmso含量为1‰。
4、培养:5%co2,37℃饱和湿度育孵箱培养72小时。
5、呈色:培养72小时吸出培养基,每孔加入100μl完全1640培养基和10μlcck8,37℃孵育4小时。
6、比色:选择620、450nm波长,在酶标仪上测定各孔光密度(od)值,记录结果。
7、同一孔450nm吸光光度值-620nm吸光光度值(背景吸光光度值)作为最终吸光度,代入以下公式。
8、细胞增殖活力(%)=[a(加药)-a(空白)]/[a(0加药)-a(空白)]×100
a(加药):具有细胞、cck溶液和药物溶液的孔的吸光度
a(空白):具有培养基和cck溶液而没有细胞的孔的吸光度
a(0加药):具有细胞、cck溶液而没有药物溶液的孔的吸光度
9、实验结果表明:在浓度为10、30μm本发明化合物的作用下,hct116细胞增殖活性受到明显抑制。
如图81所示为hct116(人结肠癌细胞)在本发明化合物(编号fb1-3-s、fb1-3-x)不同浓度下的存活率,以浓度为横坐标,存活率为纵坐标绘制的浓度-存活率曲线。从曲线上可以获知,本发明化合物(fb1-3-s、fb1-3-x)对hct116(人结肠癌细胞)的半致死浓度ic50分别为11.87μm、21.99μm。
由此可见,本发明fb1-3-s、fb1-3-x对hct116(人结肠癌细胞)活性具有抑制作用,适用于抗肿瘤药物的制备应用。
- 还没有人留言评论。精彩留言会获得点赞!
- 毛栲利素衍生物的制备方法及其抗菌活性的新用图
- 6-o-脱甲基侧厚壳桂碱衍生物及其制备和抗病毒活性应用及抗癌活性的制作方法
- 一种聚乙二醇中吡啶衍生物的合成方法
- 棉酚衍生物和它们的制备,在农药上的应用及抗癌活性的制作方法
- 用于治疗或预防神经精神病疾病的含有黄酮-6-c-葡萄糖衍生物作为活性成分的药物组合物的制作方法
- 具有抗癌活性的6-氧代-1,6-二氢-哒嗪衍生物与吉非替尼的组合的制作方法
- 2,3′-脱水-2′-脱氧-5-氟尿苷衍生物与细胞毒活性、制备方法及应用
- 2,3′-脱水-2′-脱氧-5-氟尿苷衍生物与细胞毒活性、制备方法及应用
- 具有抗肿瘤活性的哌啶酮衍生物及其制备方法
- 一种支化聚乙二醇的脂质衍生物及其组成的脂质膜结构体的制作方法