一种氯霉素半抗原及其制备方法和应用与流程
本发明涉及一种半抗原及其制备方法和应用,尤其涉及一种氯霉素半抗原及其制备方法和应用。
背景技术:
氯霉素(Chloramphenicol,CAP)是Ehrlich(1947)分离出来的一种广谱抗生素。由于其低廉的价格和稳定的抗菌性,曾在一段时间内作为饲料添加剂和兽医临床常用药品。但氯霉素有严重的毒副作用,能引起再生障碍性贫血症和婴儿灰色综合症。美国和欧共体已禁止在动物中使用氯霉素,并规定在动物源食品中不得检出,且这种不得检出的低限已达到0.01μg/kg。目前,我国动物源食品中氯霉素残留仍很严重,出口的水产品、蜂蜜屡屡被检出氯霉素残留。为保障人民的身体健康,迫切需要建立灵敏度高,特异性强,简便易行的检验方法。
氯霉素检测方法包括微生物法、色谱法、免疫测定法。微生物法易操作、费用低,但灵敏度低、特异性差。色谱法精确可靠、灵敏度高,检测极限可达到0.1μg/kg,但前处理步骤多,回收率偏低。免疫测定具有灵敏度高、特异性强、对仪器设备和人员素质要求低以及样品前处理简单等优点,适于现场监控和大规模样品筛选。
技术实现要素:
本发明的目的是提供一种氯霉素半抗原及其制备方法和应用。
本发明的第一个方面是提供一种氯霉素半抗原,其分子结构式为:
本发明的第二个方面是提供一种如本发明第一个方面所述的氯霉素半抗原的制备方法,包括以下步骤:步骤1,取氯霉素,加入叔丁基二甲基氯硅烷(TBSCl)、咪唑和无水N,N-二甲基甲酰胺(DMF),室温反应完全,纯化,得到TBS-氯霉素,其分子结构式如式(2)所示,其中氯霉素、TBSCl、咪唑和无水DMF的用量比为:1g︰700-800mg︰300-500mg︰3-7mL;步骤2,取TBS-氯霉素,加入二氯甲烷溶解,加入丁二酸酐、4-二甲氨基吡啶和三乙胺,室温反应完全,纯化,得到含琥珀酰基-TBS-氯霉素,其分子结构式如式(3)所示,其中TBS-氯霉素、丁二酸酐、4-二甲氨基吡啶和三乙胺的用量比为:0.9746g︰0.81-0.83g︰0.24-0.26g︰100-300μL;;步骤3,取含琥珀酰基-TBS-氯霉素,加入吡啶、乙腈和40%HF,冰浴冷却后,室温反应完全,纯化,得到权利要求1所述的氯霉素半抗原,其中含琥珀酰基-TBS-氯霉素、吡啶、乙腈和40%HF的用量比为:954.94mg︰2-4mL︰5-7mL︰1.5-2mL;;
优选地,上述步骤(1)中,氯霉素、TBSCl、咪唑和无水DMF的用量比为:1g︰750mg︰400mg︰5mL。
优选地,上述步骤(2)中TBS-氯霉素、丁二酸酐、4-二甲氨基吡啶和三乙胺的用量比为:0.9746g︰0.8236g︰0.2514g︰200μL。
优选地,上述步骤(3)中含琥珀酰基-TBS-氯霉素、吡啶、乙腈和40%HF的用量比为:954.94mg︰3mL︰6mL︰1.780mL。
本发明的第三个方面是提供一种氯霉素抗原,由本发明第一个方面所述的氯霉素半抗原与载体蛋白偶联得到,其分子结构式为:
本发明的第四个方面是提供一种制备如本发明第三个方面所述的氯霉素抗原的方法,包括以下步骤:步骤a,取权利要求1所述的氯霉素半抗原,加入无水二甲基甲酰胺混匀后,加入二环己基碳二亚胺和N-羟基琥珀酰亚胺,室温反应过夜,其中,氯霉素半抗原、二环己基碳二亚胺和N-羟基琥珀酰亚胺的用量比为:0.09mmol︰0.09-0.15mmol︰0.09-0.15mmol;步骤b,离心,取溶液,将溶液缓慢加入载体蛋白溶液中,所用载体蛋白的量按照半抗原与载体蛋白的摩尔比15-40︰1计;步骤c,反应完全后,透析,得到氯霉素抗原。
优选地,上述步骤a中,氯霉素半抗原、二环己基碳二亚胺和N-羟基琥珀酰亚胺的用量比为:0.09mmol︰0.1mmol︰0.1mmol。
本发明的第五个方面是提供一种氯霉素抗体,由本发明第三个方面所述的氯霉素抗原免疫动物制备得到。
本发明的第六个方面是提供由本发明第五个方面所述的氯霉素抗体制备的氯霉素酶联免疫试剂盒。
本发明的第七个方面是提供本发明第一个方面所述的氯霉素半抗原或本发明第三个方面所述的氯霉素抗原在制备氯霉素抗体中的应用。
本发明的第八个方面是提供本发明第五个方面所述的氯霉素抗体或本发明第六个方面所述的氯霉素酶联免疫试剂盒在检测动物源性食品中氯霉素残留的应用。
本发明提供的氯霉素半抗原既最大程度地保留了氯霉素的化学结构,又通过化学合成改造引入了可以与蛋白质偶联的-COOH,合成方法简单,纯度、产率较高;用该半抗原作为原料,制备适于动物免疫的抗原体系免疫动物,所得抗体的效价、特异性、亲和力都比较好;所得的抗体用于酶联免疫试剂盒,使用方便、检测成本低、检测方法高效、准确、快速、可同时检测大批量的样本,适于动物源性食品中氯霉素残留的现场监控和大量样本的筛查。本发明的氯霉素半抗原在氯霉素的检测中发挥重要作用。
附图说明
图1为氯霉素半抗原合成路线图。
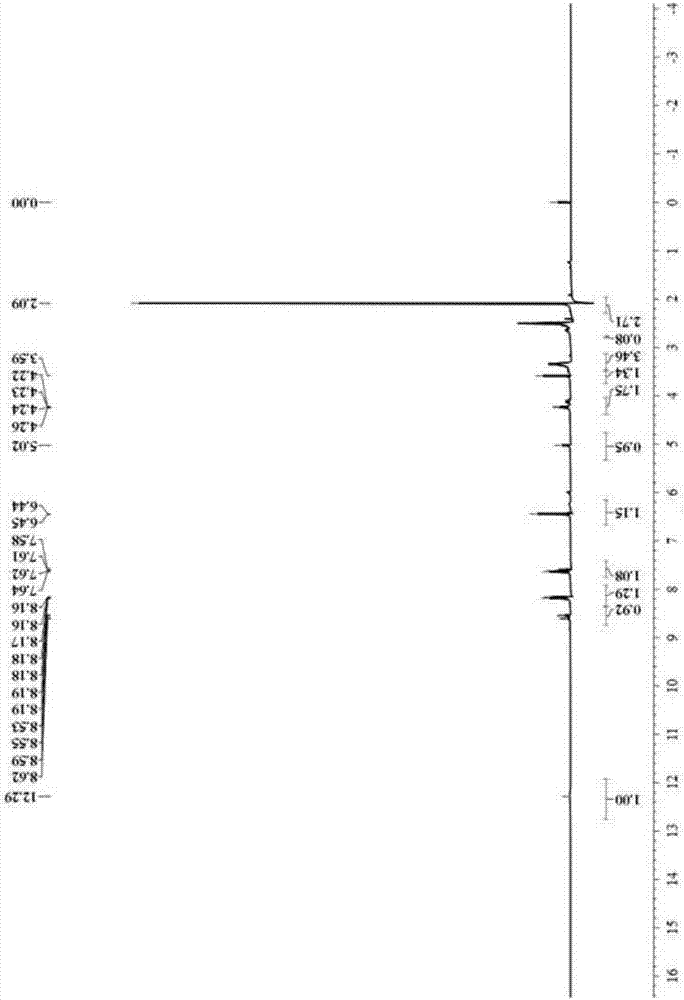
图2为氯霉素半抗原核磁共振氢谱图。
图3为氯霉素ELISA标准曲线。
具体实施方式
下面结合具体的实施例来进一步阐述本发明。应理解,这些实施例仅用于说明本发明,而不用来限制本发明的范围。
实施例1:氯霉素半抗原的合成
步骤1:
1、称取1g氯霉素于100mL的圆底玻璃瓶中,往其中分别加入750mg叔丁基二甲基氯硅烷(TBS-Cl)、400mg咪唑和5mL无水二甲基甲酰胺(DMF)。选择合适大小的小磁石置于圆底玻璃瓶中,用锡箔纸封好瓶口。将圆底玻璃瓶放在磁力搅拌器上,调节适宜的转数,室温搅拌。同时用薄层色谱法(TLC)跟踪原料反应完全,此反应可在1h内反应完全,且此过程的最适展开剂为乙酸乙酯:正己烷:乙酸=7:3:0.1。
2、薄层色谱法监控反应完全后,用量筒量取50mL乙酸乙酯稀释反应液,将稀释后的反应液转移至分液漏斗中。往分液漏斗中加入10mL 0.1M HCl,手动摇晃5min,将其固定在铁架台上静置10min,释放下层溶液于小烧杯中,上层有机相留在分液漏斗。同样,用10mL饱和NaCO3和10mL饱和NaCl对分液漏斗中的有机相进行依次洗涤。经三次洗涤后,将上层有机相转移至锥形瓶中,烧杯中的水相转移至分液漏斗,用20mL乙酸乙酯对其进行反提,弃去下层,合并上层有机相于锥形瓶。往锥形瓶中加入适量无水NaSO4进行干燥,无水NaSO4结块即可。
3、取干净的200mL的圆底玻璃瓶,称量可得瓶净重163.7041g。将锥形瓶中的有机相转移至该圆底玻璃瓶,用少量的乙酸乙酯多次润洗锥形瓶,并将润洗液合并到圆底玻璃瓶中置于旋转蒸发仪上旋干(水浴温度为40℃,转数60),未完全旋干则用氮吹仪氮吹至完全干燥。称量可得圆底玻璃瓶和反应产物重165.3077g。其中反应产物(含TBS-氯霉素)的质量为1.6036g。
4、纯化反应产物。往圆底玻璃瓶中加入8mL甲醇溶液溶解样品,待完全溶解后吸450μL于2mL离心管中,再往离心管中加入约1500μL的甲醇溶液,混匀后进行跑大板纯化。此过程使用的展开剂为乙酸乙酯:正己烷:乙酸=70:30:1。跑完大板后,刮下含药品的硅胶粉部分,碾碎,置于烧杯中。往烧杯中加入适宜的乙酸乙酯搅拌,放在超声仪上超声10min,同样的步骤,直至洗下所有的药品(通过点小板来检测是否还有药品)。用滤膜过滤所有的洗脱液于圆底玻璃瓶,空瓶重:163.6041g,将圆底玻璃瓶置于旋转蒸发仪进行旋干。旋干后得到的圆底玻璃瓶重(含药品):164.5787g。此时,纯化后的反应产物的量为0.9746g。
步骤2:
1、用4mL二氯甲烷溶解上述步骤所得的产物,吸取303.7μL于2mL离心管中,用氮气吹干,封口,置于4℃保存。圆底玻璃瓶所剩的900mg转移至新的100mL的圆底玻璃瓶中,瓶净重为:111.4161g,再用6mL二氯甲烷分批次润洗干净转移。往圆底玻璃瓶中分别加入0.8236g丁二酸酐、0.2514g 4-二甲氨基吡啶(DMAP)、200μL三乙胺。选择合适大小的小磁石置于圆底玻璃瓶中,用锡箔纸封好瓶口。将圆底玻璃瓶放在磁力搅拌器上,调节适宜的转数,室温搅拌。同时用薄层色谱法(TLC)跟踪原料反应完全,此反应约在6h内反应完全,且此过程的最适展开剂为乙酸乙酯:二氯甲烷:乙酸=6:4:0.1。
2、薄层色谱法监控反应完全后,用量筒量取50mL乙酸乙酯稀释反应液,将稀释后的反应液转移至分液漏斗中。往分液漏斗中加入10mL饱和NH4Cl,手动摇晃5min,将其固定在铁架台上静置10min,释放下层溶液于小烧杯中,上层有机相留在分液漏斗。同样,再用10mL饱和NH4Cl对分液漏斗中的有机相再一次洗涤。经两次洗涤后,将上层有机相转移至锥形瓶中,烧杯中的水相转移至分液漏斗中,用20mL乙酸乙酯对其进行反提,弃去下层,合并上层有机相于锥形瓶中。往锥形瓶中加入适量无水NaSO4进行干燥,无水NaSO4结块即可。
3、取干净的200mL圆底玻璃瓶,称量可得瓶净重163.6046g。将锥形瓶中的有机相转移至该圆底玻璃瓶中,用少量的乙酸乙酯多次润洗锥形瓶,并将润洗液合并到圆底玻璃瓶中置于旋转蒸发仪上旋干(水浴温度为40℃,转数60),未完全旋干则用氮吹仪氮吹至完全干燥。称量可得圆底玻璃瓶(含反应产物琥珀酰基-TBS-氯霉素)重164.6587g。其中反应产物(含琥珀酰基-TBS-氯霉素)的量为1.0541g。
步骤3:
1、用2mL甲醇溶液溶解样品,吸取100μL于2mL离心管中,用氮气吹干,封口,置于4℃保存。剩余部分样品置于旋转蒸发仪上旋干,称量得药品重量为954.94mg。往样品中分别加入3mL吡啶、6mL乙腈、1.780mL 40%HF,整个加样过程,圆底玻璃瓶应置于冰盒中,冰浴冷却,。待冷却后将圆底玻璃瓶置于磁力搅拌器上,放入适宜大小的磁力搅拌子,调节适宜转数,室温搅拌过夜。转天早晨用薄层色谱法(TLC)跟踪原料反应完全,此反应约在9.5h内反应完全,且此过程的最适展开剂为乙酸乙酯:二氯甲烷:乙酸=7:3:0.1。
2、薄层色谱法监控反应完全后,用量筒量取50mL乙酸乙酯稀释反应液,将稀释后的反应液转移至分液漏斗中。往分液漏斗中加入10mL 0.1M HCl,手动摇晃5min,将其固定在铁架台上静置10min,释放下层溶液于小烧杯中,上层有机相留在分液漏斗。同样,再用10mL 0.1M HCl、10mL饱和NaCl、10mL饱和NaCl对分液漏斗中的有机相进行依次洗涤。经四次洗涤后,将上层有机相转移至锥形瓶中,烧杯中的水相转移至分液漏斗中,用20mL乙酸乙酯对其进行反提,弃去下层,合并上层有机相于锥形瓶中。往锥形瓶中加入适量无水NaSO4进行干燥,无水NaSO4结块即可。
3、取干净的圆底玻璃瓶,称量可得瓶净重163.5687g。将锥形瓶中的有机相转移至该圆底玻璃瓶中,用少量的乙酸乙酯多次润洗锥形瓶,并将润洗液合并到圆底玻璃瓶中置于旋转蒸发仪上旋干(水浴温度为40℃,转数60),未完全旋干则用氮吹仪氮吹至完全干燥。称量可得圆底玻璃瓶(含反应产物琥珀酰基-氯霉素)重164.1727g。其中反应产物(含琥珀酰基-氯霉素)的量为0.604g。
4、因为产物不纯,含两种物质,所以通过使用柱层析技术将不同组分分离开。湿法装柱,硅胶高度约为30cm,硅胶上层装入约0.5cm的无水硫酸钠,防止添加溶剂时使得样品层不再整齐。装柱完备后,在柱子上端使用加压泵进行加压,使得柱子更结实。再用展开剂(乙酸乙酯:二氯甲烷:乙酸=7:3:0.1)“走柱子”。将样品用少量的展开剂溶解后上样。上样完毕接着用同样的展开剂洗脱,用锥形瓶接洗脱液,将不同组分分离开,分离的过程通过点小板来确定样品是否洗脱及分别收集。将收集好的样品过滤,除去一同洗下的硅胶后,转移至干净的圆底玻璃瓶,旋干并称重。最先洗脱出的物质,旋干并氮吹后所得质量为351.9mg。后洗脱出的产物,旋干并氮吹后所得质量为280.6mg。
实施例2:氯霉素半抗原的鉴定
取上述实施例1的产物经核磁共振氢谱测定,如图2所示,核磁共振数据如下,说明半抗原合成成功:
1H NMR(400MHz,DMSO)δ12.29(s,1H),8.57(dd,J=25.4,8.9Hz,1H),8.35-7.91(m,1H),7.61(dd,J=14.3,8.8Hz,1H),6.45(d,J=3.6Hz,1H),5.02(s,1H),4.24(dd,J=7.5,4.9Hz,2H),3.59(s,1H),3.49-3.13(m,3H),2.76(s,1H),2.09(s,3H)。
实施例3:氯霉素抗原
向0.09mmol所述琥珀酰基氯霉素半抗原中加入0.6ml的无水DMF,混匀后,加入0.1mmol的DCC和0.1mmol的NHS活化半抗原上的羧基基团,搅拌反应过夜后,离心去沉淀,将溶液平均分为三份,分别逐滴加入到不同的载体蛋白溶液中,所述载体蛋白为BSA,OVA,KLH等,所述溶解蛋白的缓冲液为碳酸盐缓冲液或硼酸盐缓冲液,所用蛋白的量按照半抗原与蛋白的摩尔比15-40︰1计算。待羧基活化的半抗原与蛋白分子上的氨基偶联反应过夜后,将反应液转移到半透膜中,使用10mM的pH7.2的PBS缓冲液于4℃透析3-5天,得到包被原,经紫外鉴定,氯霉素半抗原与载体蛋白偶联成功。将透析完毕的溶液按照所用载体蛋白的量用PBS稀释成1mg/ml。
实施例4:抗血清制备
将1mg/ml的氯霉素-KLH或氯霉素-BSA作为免疫原与等体积的弗氏佐剂混合搅拌乳化,首次免疫用弗氏完全佐剂,以后免疫使用弗氏不完全佐剂,每间隔10至15天对小鼠免疫一次,第三次免疫后小鼠眼眶采血检测血清效价和抗体特异性。
实施例5:血清效价及抗体特异性检测
将1mg/ml的氯霉素-OVA包被原使用碳酸盐缓冲液稀释合适的倍数后,加入到96孔酶标板中于37℃包被3小时,将氯霉素标准品使用样品稀释液(PBS中含有0.1%的吐温20和明胶)稀释后,每孔加入50ul,以样品稀释液作为非抑制孔对照,随后加入经过样品稀释液稀释合适倍数的血清,于37℃温浴30min后,洗脱,再加入羊抗鼠酶标二抗反应30min,洗脱酶标二抗后,加入含有OPD或TMB的底物缓冲液显色5-10min,于酶标仪检测OD值。
本发明以氯霉素-OVA作为包被原,氯霉素-KLH作为免疫原所获得的抗血清,所建立的ELISA标准曲线如图3所示,抑制中浓度为9ng/ml。
以上对本发明的具体实施例进行了详细描述,但其只是作为范例,本发明并不限制于以上描述的具体实施例。对于本领域技术人员而言,任何对本发明进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!