巴斯德毕赤酵母中人RANK胞外段蛋白的表达纯化及其应用的制作方法
本发明涉及蛋白的表达纯化,更具体地,涉及巴斯德毕赤酵母中人rank胞外段蛋白的表达纯化。
背景技术:
1.1rank
tnf和tnfr超家族具有功能多样性,tnf超家族成员在一系列组织和器官中都有表达,通常在免疫细胞表面有表达,近年来对tnf/tnfr的研究揭示了免疫系统和其他生物学系统之间的关系,以及许多疾病的调控网络。nf-κb的受体活化因子rank,或称为tnf相关活化诱导的细胞因子受体(trance-r)[1]、破骨细胞分化激活受体(odar)[2]是rankl的信号受体。rank(tnfrsf11a)属于肿瘤坏死因子受体家族中的一员与rankl(rank配体)、opg(骨保护素)共同组成骨代谢稳态的基础,其中opg/rankl的比率变化影响着破骨细胞的发育及活化。rank是一种i型三聚跨膜蛋白,胞外段蛋白富含4个同型半胱氨酸假重复序列与大鼠具有70%的同源性。人的rank基因由616个氨基酸残基构成,c端胞内区没有内源酶活性,需要n端(胞外段)与三聚体rankl(nf-κb受体激活剂配体)结合后引发自身三聚化,招募下游的接合体和对接蛋白,从而激活下游信号开始细胞内的信号级联传导。主要激活nf-κb和jnk通路[5]。rank基因缺陷性小鼠中因为缺乏破骨细胞而表现为骨硬化症及淋巴细胞缺失、淋巴结的发育不良。研究发现rank-rankl-opg系统对骨生成稳态、淋巴结的形成、淋巴细胞的发育、乳腺发育、m细胞的发育、树突状细胞(dc)的存活、中枢免疫耐受的建立、血管钙化、体温调节等生理过程发挥着重要调节作用[5-8]。t细胞表达的rankl可以促进t细胞增殖,同时t细胞分泌ifn-γ又促使traf6蛋白酶体降解,进而抑制了rank信号[9,10]。这暗示着rank-rankl-opg系统是骨与免疫系统之间相互关联的重要介质。骨免疫作用机制的发现为骨质疏松症、类风湿关节炎、骨转移性肿瘤等骨破坏性疾病提供一个新的思路与治疗手段。
然而rank/rankl/opg并不局限于骨及免疫系统,其在动脉粥样硬化及糖尿病中也扮演着重要角色。在动脉粥样硬化中rankl/rank可以直接促进血管平滑肌的钙化,或者通过分泌il-6和tnfa增强巨噬细胞的旁分泌间接促进血管平滑肌的钙化,其中opg可预防血管钙化。关于糖尿病的研究指出在i型糖尿病小鼠模型中,rankl参与的调节性t细胞可以保护β胰岛细胞免受细胞毒性破坏。最近研究在临床水平上揭示了血清中高水平的可溶性肝rankl与2型糖尿病(t2dm)风险正相关。
此外,胸腺中枢耐受的建立是为了防止自身免疫性疾病,但在对肿瘤患者的免疫治疗中,解除患者的免疫耐受状态是个新的治疗思路。最近的一项研究表明通过瞬时阻断rankl可以打破中枢耐受,进而增加肿瘤特异性效应t细胞发挥抗肿瘤作用。
rank可以广泛表达于胸腺、肝、结肠、乳腺、前列腺、胰腺、骨髓、心脏、肺、脑、骨骼肌、肾、肝和皮肤[26]。在本实验室前期研究中通过动物成瘤实验发现,rank-fc对结直肠癌具有抑制作用。这为我们利用人源化结直肠癌小鼠模型对rank胞外段蛋白进行体内抑制肿瘤生长的功能验证奠定了实验基础。另外,因哺乳动物中的表达量低及及大肠杆菌中的纯化困难,而巴斯德毕赤酵母表达系统作为目前最成功的外源蛋白表达系统之一,与大肠杆菌和哺乳动物细胞表达系统相比具有使用简单、表达量高、蛋白便于纯化、接近人源蛋白糖基化等优点,因此我们利用此系统大量表达人源rank胞外段蛋白(以下简称rank-n)。
本研究中通过rank受体胞外段蛋白来实现阻断rankl-rank信号通路,这种重组蛋白能结合rankl而自己本身由于没有细胞内结构不能传递下游信号,这样可以阻断rankl-rank信号。虽然已经有人使用rank-fc融合蛋白阻断该信号通路,但是其表达量和蛋白活性都不是很高。我们这里表达的rank-n相比rank-fc融合蛋白具有更高的蛋白表达量,同时避免了复杂的蛋白与融合部分的分离操作,增加了产品的回收率。本次研究构建了rank-n巴斯德毕赤酵母表达菌株,并建立超滤-sephadexg-50凝胶过滤层析-qsepharoseff离子交换层析三步纯化方法,得到具有功能活性,纯度超过95%的可溶性的人rank-n蛋白。体外体内实验均证明其具有阻断rankl-rank信号的功能,为进一步研究rankl-rank信号通路的科研和临床应用奠定了基础。
1.2巴斯德毕赤酵母表达系统
巴斯德毕赤酵母是一甲醇营养型巴斯德毕赤酵母,能够利用甲醇作为唯一碳源。它兼具原核表达系统成本低廉、操作简易、表达量高及真核生物表达系统对外源蛋白翻译后进行加工修饰等优点。因此其在生物制药业中受到越来越多的关注已被广泛应用于外源蛋白的表达。巴斯德毕赤酵母能代谢甲醇源于过氧化物酶中的醇氧化酶1(aox1)基因,aox1启动子是目前已知的最强及最严格的调节真核启动子之一。巴斯德毕赤酵母有两个醇氧化酶基因,aox1和aox2,其中aox1比aox2被更强烈地转录[27]。目前,越来越多的蛋白质已使用此启动子表达,其产率高达14.8克/l[28],优化条件后可达重组蛋白的20-30克/l[29]。aox1启动子在葡萄糖或甘油的存在下被抑制,但在甲醇的存在下会强烈上调。aox1启动子能够驱动aox1蛋白质或其它重组蛋白质以非常高的水平表达,达到总可溶性蛋白的表达30%以上[30]。
巴斯德毕赤酵母通常以营养单倍体状态存在,当有氮限制时会导致交配并形成双倍体细胞[31]。所有巴斯德毕赤酵母表达菌株源自野生型菌株nrrl-y11430。营养缺陷型突变体(gs115)、蛋白酶缺陷型菌株(smd1163,smd1165,smd1168)是最常被使用的表达菌株。从甲醇的利用率可将巴斯德毕赤酵母可分为三种表型,即mut+(甲醇利用快型),muts(甲醇利用慢型),mut-(甲醇不利用型)[32]。ppic9k是一种分泌型表达载体,它通过酿酒酵母α-信号肽,使外源蛋白质分泌入培养液中,其中目的蛋白可占分泌总蛋白的50%以上且自身分泌的蛋白少易于下游靶蛋白的分离纯化[30,33-35]。anandghosalkar[36]等发现在gs115菌株使用α-mf信号肽(肽酿酒酵母α交配因子前导肽),蛋白表达量可高达200mg/l。
巴斯德毕赤酵母表达系统有以下优点:
(1)具有快速生长速率且易于高密度发酵;(2)发酵培养基成本低廉,表达量高,外源基因的表达水平比原核细胞及酿酒酵母高,多数>1g/l。(3)应用分泌型表达载体通过α-信号肽可使表达产物分泌到培养基中便于靶蛋白的分离纯化。(3)无内毒素和噬菌体的污染;(4)有易于基因操纵的酵母表达载体;(5)针对巴斯德毕赤酵母的裂解病毒无人类致病性;(6)具有多肽折叠,糖基化,甲基化,酰基化,蛋白剪切等翻译后修饰功能(7)遗传背景清楚,遗传性状稳定[37]。
巴斯德毕赤酵母表达系统的缺点:
(1)发酵周期较长,容易污染,应及时通过sds-page和考马斯亮蓝染色鉴定缩短时间避免菌体蛋白干扰。
(2)甲醇易燃易爆具有细胞毒性,大量堆放存在潜在危险。
(3)蛋白酶降解经常影响着巴斯德毕赤酵母外源蛋白的成功分泌[38-40]。
(4)高度甘露糖型糖基化:巴斯德毕赤酵母表达系统较酿酒酵母是最接近人源蛋白糖基化的表达系统[41]。酵母执行的翻译后修饰如n-和o-糖基化对保证重组蛋白的生物学活性是必不可少的[41]。然而,酵母菌的n-多聚糖是异质高甘露糖型糖基化。当高甘露糖型n-多聚糖从人血液中清除时可以引起免疫原性反应。目前的解决策略主要集中以下两种方法:1.糖基化位点的定点突变[42];2.糖基化的基因工程改造[43][44]。
1.3rank/rankl/opg简介
1.3.1rankl-rank-opg系统
nf-κb受体激活剂配体(rankl)
rankl最早是由四个不同的研究组独立发现的,并分别称之为rankl,trance,odf和opgl,现在也统一命名为tnfsf[11]。rankl属于tnf超家族成员,是一种二型跨膜蛋白,与tnf配体超家族成员如trail,fasl以及tnfa具有明显的同源性,人与大鼠的rankl的同源性高达83%,说明rankl在进化过程中是高度保守的[45]。rankl可以以膜结合或者游离的方式存在,其中游离形式的rankl由金属蛋白酶-解偶联蛋白tnfα转化酶(tace)对膜结合形式的rankl的剪切而产生的[7]。进一步研究显示rankl在体内有三种同源异构体,其中最短的同源异构体缺少胞内区和跨膜区并且可能具有抑制性作用,这与游离形式的rankl在介导破骨细胞生成过程中的效率更低发现一致[46]。rankl在多种组织细胞中都有表达,包括t淋巴细胞[45]、成骨细胞、骨基质细胞和肺上皮细胞[47]。在发育过程中,小鼠胚胎的脑、心脏、肾脏、骨骼肌和皮肤中均可以检测到ranklmrna存在,另外,小鼠胚胎15天的软骨细胞中也检测到大量的ranklmrna存在。许多细胞因子或者激素如糖皮质激素、维生素d347、il-1、tnf-a、tgf-b、wnt配体[48]和lps[49]可以高效诱导rankl的表达。
nf-κb受体激活剂(rank)
rank是rankl的信号受体,也被称为trance-r和odar,现在也被统一命名为tnfrsf11a。rank属于tnf受体超家族成员,是一种一型三聚体跨膜蛋白,与cd40胞外域部分具有高度的同源性,人与大鼠的rank同源性高达70%,在进化上高度保守。人的rank由616个氨基酸组成,n端胞外区含有4个同型半胱氨酸假重复序列,跨膜区很短,c端胞内区没有内源酶活性,需要n端的胞外区与三聚体rankl结合后引发自身三聚化,从而招募下游的接合体和tnfr相关因子(tnfreceptorassociatedfactors,trafs)中的traf2,3,5和6,接头/支架蛋白gab2和泛素连接酶cbl,激活胞质内的信号级联反应,最终激活jnk2/stat5通路和nf-κb通路[50]。rank可以广泛表达于胸腺、肝、结肠、乳腺、前列腺、胰腺、骨髓、心脏、肺、脑、骨骼肌、肾、肝和皮肤。
骨保护素(opg)
骨保护素是在尝试靶向大鼠tnfrsf同源蛋白的探索中首次发现的,也被称之为破骨细胞形成抑制因子(ocif),tr1,或滤泡树突状细胞源性受体-1(fdcr-1),现在被统一命名为tnfr11b。opg属于tnf受体超家族成员,是一种分泌型糖蛋白,结构上具有tnf受体超家族特征性的半胱氨酸假重复序列,人与小鼠的opg的同源性高达85%,进化上高度保守[51]。全长的opg蛋白由401个氨基酸组成,n端含有21个氨基酸组成的信号肽,经过糖基化后,成熟的opg含有380个氨基酸,整个蛋白含有7个结构域:4个富含半胱氨酸的n末端结构域(结构域1-4),两个死亡域同源区(域5和6)和一个c-末端的肝素结合域多肽区(结构域7)[17,52]。
研究发现体内多种组织如皮肤,肝,肺,和成年小鼠的心脏均可以分泌opg,并且胚胎期7天和15天的胚胎组织中表达的opg处于峰值[51]。opg主要在骨基质细胞上表达,但也可以在树突状细胞,和滤泡树突状细胞,b淋巴细胞中被诱导表达[53]。opg的表达可以被一系列维持骨稳态的因子正向的或者负向的调控[54]。例如tgf-b,il-1,tnf,雌激素和wnt配体可以诱导opg的表达,而前列腺素e2(pge2)和糖皮质激素则抑制opg的表达[54]。
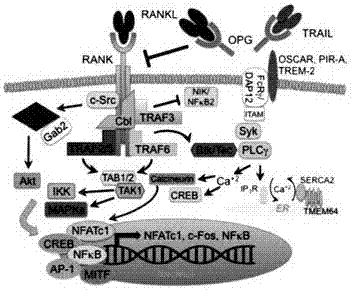
rank信号通路如图1所示。rank受体缺乏内源酶活性,因此,利用接合体和对接蛋白的相互作用去激活下游信号,包括trafs2,3,5和6,接头/支架蛋白gab2和泛素连接酶cbl。gab2和cbl与rank介导激活的蛋白酪氨酸激酶(c-src)、3-磷酸肌醇激酶(pi3k)和蛋白激酶bγ(akt3)依次相结合,而肿瘤坏死因子受体相关因子trafs2和6可以激活tab1/tab2/tak1复合物,这些(连同其他上游激酶)激活iκb激酶β(ikkβ)和丝裂原活化蛋白激酶(mapk)。这些途径的激活促进了转录因子的移位和活化,包括破骨细胞活化t细胞核因子l(nfatc1)、转录增强因子creb、nfκb、ap-1和mitf。另外,在破骨细胞生成中重要的介导物质dap12、fcrγ(免疫球蛋白样受体)与细胞表面受体oscar,pir-a及trem-2协同激活syk-plcγ通路和钙通道。walshmc,choiy.frontimmunol.2014oct20;5:511.
1.3.2rankl-rank信号在发育过程中的作用
rank-rankl信号在多种组织的上皮细胞发育和分化过程发挥着重要的调节作用,这种rank依赖的调控机制不但影响器官发育过程,也影响着免疫系统的功能和癌症的发生。
rank和rankl基因缺陷小鼠表现为二级淋巴组织发育缺陷,如淋巴结,派尔集合淋巴结,肠粘膜的微小肠道隐窝斑以及脾脏[55,56]。rank和rankl基因缺陷小鼠的乳腺组织发育也存在着缺陷。乳腺组织的发育需要通过rank激活下游ikka依赖的非经典nf-κb信号通路[57]。微皱细胞(m细胞)来源于肠道黏膜上皮细胞,是粘膜免疫中的一种抗原呈递细胞,可诱导免疫黏膜免疫应答或免疫耐受。rankl信号在m细胞发育过程中非常关键。rankl基因缺陷小鼠中m细胞发育分化缺陷[7]。
rankl-rank信号在胸腺器官发育中扮演着非常关键的角色,是尤其在胸腺髓质上皮细胞的发育和分化过程中发挥着极其重要的调控作用,进而影响发育中的t细胞进行阴性选择[8,58]。定位于胸腺髓质区的上皮细胞被称为髓胸腺上皮细胞(mtecs),其来源于非造血细胞,胸腺髓质上皮细胞通过消除那些潜在表达的自身反应性tcr的t细胞和产生具有免疫抑制性质的foxp3阳性调节性t细胞(treg细胞)从而建立中枢免疫耐受[8,59]。在缺少胸腺细胞提供的rankl时mtecs数目和比例显著减少,这个过程似乎在发生在新生儿期,在tcrαβt细胞未产生之前,tcrγδt细胞表达的rankl就可以提供mtec前体细胞发育分化的所需要的信号[60]。mtecs的发育分化需要tnf受体超家族成员ltβr、cd40和rank依赖的信号通路,以便诱导自身免疫调节因子(aire)和组织特异性抗原(tsas)的充分表达,但这些受体的下游激活的通路是否完全一致目前尚不清楚。traf3基因缺陷鼠提示ltβr和cd40在激活非经典的nf-κb通路上发挥着相似的作用,但rank在mtecs发育中显然还提供了额外的必须信号[61]。遗传学实验显示rank信号是通过下游的traf6分子介导的[58]。总之,这些数据表明rank介导的信号在免疫调节和免疫稳态的发育过程中是必需的。
1.3.3rankl-rank-opg与骨代谢稳态
骨代谢包括骨形成与骨吸收,骨形成与骨吸收之间只有处于动态平衡,骨组织才能具有正常的形态,发挥应有的功能。骨代谢稳态是主要由两种细胞维持的:成骨细胞先形成类骨物质并矿化形成新骨,破骨细胞清除旧骨的矿物质。骨代谢稳态过程是由成骨细胞和破骨细胞共同介导的,许多细胞因子、激素、以及生长因子均参与其中。在体外,可以通过破骨细胞/骨髓基质细胞共培养得到破骨细胞,需细胞-细胞接触依赖性培养,rankl-rank信号可以激活这个发育过程[62,63]。巨噬细胞集落刺激因子m-csf可以刺激破骨前体细胞的增殖以及诱导rank及rank信号下游的转录因子c-fos,nfatc1/nfatc2以及nf-κb的家族成员p50和p52的表达从而促进破骨细胞的发育[64,65]。rankl基因缺陷鼠由于破骨细胞本身正常的发育而表现为严重的骨质疏松症以及牙齿发育不全[66]。
1.3.4rankl-rank-opg系统与肿瘤的关系
rankl-rank-opg系统在众多破骨细胞依赖的骨转移癌症病理过程中都发挥重要的调节作用。癌细胞骨转移后以及其产生的肿瘤相关因子加速了骨吸收的过程,并且释放了一些生长因子促进了癌细胞的增殖。许多研究发现,通过阻断rankl来抑制破骨细胞的发育可以抑制骨癌组织边缘癌细胞增殖并诱导其凋亡。这一现象在前列腺癌[67],乳腺癌[68],骨髓瘤[69]的小鼠动物模型中都已被证实。然而,rankl-rank-opg系统也可以通过破骨细胞非依赖的途径参与肿瘤的发生发展,前列腺癌,乳腺癌,肺癌,肾癌,黑色素瘤的癌细胞都有rank的表达,重要的是,对一系列实体瘤分析发现原发的肿瘤细胞和骨转移的肿瘤细胞上rank的表达并没有显著的不同,说明rankl-rank-opg系统在肿瘤发生发展中起着多方面复杂的作用[70]。
rankl,rank以及opg在前列腺癌组织中表达水平是增高的,而正常组织中这三种蛋白表达水平是很低的。rankl,rank以及opg的表达高低与前列腺癌的临床病理组织指标是正相关的,即高水平的rankl,rank以及opg显示着肿瘤的恶性程度更高[71]。另外,在前列腺癌细胞中,rankl可以激活ikka以及抑制转移抑制基因maspin,促进前列腺癌的发生发展[72]。
rankl/rank系统是lobulo-alveolar区乳腺组织发育所必需的。rankl和rank基因缺陷鼠不能分泌乳液,并且乳腺小泡上皮细胞不能正常的增殖[73]。在小鼠的乳腺癌模型中,rank信号过度激活促进乳腺癌的发生,而抑制rank信号可以抑制乳腺癌的发生发展[74]。
1.3.5以rankl作为治疗靶点
由于rankl在破骨细胞的分化和激活中扮演了至关重要的角色,以及可作为溶骨性疾病发展过程的中介因子,使得rankl成为一个潜在应用的药物作用靶点[75]。目前,已经开发了不同种类的rankl阻断剂在体内来阻断rankl,如fc-opg、rank-fc和opg模拟多肽。fc-opg和rank-fc分别由opg和rank的胞外段与fc片段融合而成,重组人源和鼠源的fc-opg和rank-fc已被成功研制出来。鼠源rank-fc融合蛋白已被证明既能与鼠rankl结合也能与人rankl结合,却不能与肿瘤坏死因子相关凋亡诱导因子(trail)结合[15,16]。鉴于鼠源性单克隆抗体的多次给药易出现人抗鼠抗体的情况,目前人源单克隆抗体是进行临床治疗的主要发展趋势。
rank/rankl信号通路在癌症的发生发展过程中所扮演的角色还不是很清楚,但是,最近的研究已经证明rank/rankl信号通路的激活可以促进孕激素诱导的乳腺上皮细胞增殖以及癌变,抑制rank信号可以阻断乳腺癌细胞生长以及乳腺癌细胞肺转移[74]。因此,阻断rank/rankl/opg信号通路可能有抗肿瘤的作用。在小鼠模型中,opg可以抑制骨溶解相关的骨疾病同时也能缩小病灶的大小,opg-fc融合蛋白也被开发为一种潜在的治疗药物,并且在临床试验中用来治疗多发性骨髓瘤,乳腺癌,前列腺癌以及胃癌等[76,77]。虽然opg-fc在减少骨吸收的方面和pamidronate的效果类似,但是它的半衰期更短,并且对rankl没有特异性,opg可以阻断trail,而trail在介导先天免疫细胞杀伤肿瘤过程中发挥着重要作用[17]。而最近的研究显示opg可以促进肿瘤细胞的存活,虽然opg作为肿瘤细胞的保护因子目前还缺乏体内的证据,但是其潜在的促进肿瘤细胞的生长和转移功能限制了它的临床研究以及应用前景[17]。
作为rankl的唯一靶受体的rank,其应用优势越来越多的受到人们关注。目前rank-fc这种融合蛋白已经被获得并开展了一系列试验,研究发现注射rank-fc后实验组小鼠骨密度相对于对照组呈剂量依赖性增加[18],微型ct显示实验组小鼠骨小梁数量的增加并伴有骨小梁厚度的降低[19]。rank-fc蛋白在多发性骨髓瘤(mm)中可限制骨质破坏,可明显减少肿瘤负荷和血清m蛋白,而在opg-fc治疗组中血清m蛋白平均降低仅为25%[20]。schmiedelbj在mm及慢性淋巴细胞白血病(cll)的治疗研究中发现相对于狄诺塞麦,rank-fc可高效激活nk细胞的抗肿瘤作用[21]。另外,在人类骨肉瘤中使用rank-fc可以抑制erk信号,并同时激活caspase-3介导的失巢凋亡抑制肿瘤[22]。研究表明,rank-fc可以显著的抑制破骨细胞的活性和骨吸收,进而阻断骨肉瘤、多发性骨髓瘤、肺癌[23]、乳腺癌[24]、前列腺癌[25]等造成的骨质破坏。
denosumlab(狄诺塞麦)是首个被fda批准的rankl抑制剂。它是一种人igg2单克隆抗体,通过阻断rankl抑制破骨细胞的功能及阻止骨形成与骨吸收。在绝经后骨质疏松症i期临床实验中对49例健康的绝经后妇女进行狄诺塞麦单剂量、随机、双盲、剂量递增的皮下注射实验研究,剂量依次为0.01,0.03,0.1,0.3,1.0或3.0毫克/kg。随后观察6个月,最高剂量的三组观察长达9个月。口服安慰剂、阿仑膦酸钠作为对照,实验显示与以前使用的阿仑膦酸钠相比,骨密度可显著增加。通过检测尿ntx发现骨吸收被抑制,经白蛋白校正后血钙浓度有一个短暂降低及血清甲状旁腺激素的瞬时升高[78]。ii、iii期临床试验阶段证明其可以降低椎骨骨折、髋部骨折和非椎骨骨折的风险。在后续的ii、iii期临床试验中狄诺塞麦的耐受性良好,没有药物相关的严重不良事件(saes)报告。用药方式为:皮下注射,每4周120毫克用于预防sre;每6个月60毫克用于预防骨质疏松症和骨损失[79]。最近的iii期临床试验显示,在非转移性雄激素抗性的前列腺癌中使用狄诺塞麦可以显著增加骨转移的生存和延迟首次骨转移时间[80]。此外,相关研究指出狄诺塞麦在骨巨细胞瘤(gctb)中已应用于ii期临床,在无法手术和严重手术并发症组中结果令人振奋,显著减少了gctb患者手术的需要[81]。目前,狄诺塞麦用于早期乳腺癌骨转移的iii期临床预防工作正在进行中(d-care,nct01077154)[82]。狄诺塞麦的平均半衰期为28天,潜在不良反应有低钙血症、感染、颚骨坏死及骨代谢抑制等。长期使用狄诺塞麦对感染及免疫抑制等方面的安全性尚有待于进一步观察[82]。
综上所述,抗rankl治疗为探索骨疾病、癌症病人的骨转移、溶骨性骨肿瘤等骨破坏性疾病的生物治疗方法提供了新思路,为临床疾病的治疗和预防带来了新的希望。
在现有技术中,主要还存在以下几个方面的缺陷:1)基因重组蛋白在哺乳动物细胞中的表达量低及及大肠杆菌中的纯化困难;2)虽然已经有人使用rank-fc融合蛋白阻断该信号通路,但是其表达量和蛋白活性都不是很高;3)鼠源rank-fc融合蛋白已被证明既能与鼠rankl结合也能与人rankl结合,却不能与肿瘤坏死因子相关凋亡诱导因子(trail)结合[15,16]。鉴于鼠源性单克隆抗体的多次给药易出现人抗鼠抗体的情况,目前人源单克隆抗体是进行临床治疗的主要发展趋势。
技术实现要素:
2.1本发明所要解决的技术问题
(1)本研究首次构建新型rankl阻断剂(rank胞外段蛋白)的巴斯德毕赤酵母(pichiapastoris),保藏于中国微生物菌种保藏管理委员会普通微生物中心(cgmcc),保藏地址:北京市朝阳区北辰西路1号院3号,编号为cgmccno.12500,于2016年05月26日保藏)表达系统,获得多拷贝阳性酵母转化菌株,建立超滤—sephadexg-50—qsepharoseff的纯化方案,并获得大量高纯度目的蛋白。表达量超过30mg/l,为以后的实验奠定了基础。
(2)成功建立人源化结直肠癌小鼠模型,发现了rank-n具有抑制结直肠癌体外生长中的功能,为rank蛋白的成药性打下了良好基础,对探索癌症新的生物治疗方法提供了新的思路。
(3)通过等离子共振(spr)技术分析rank与rankl蛋白相互作用,测定两者的亲和解离常数kd为2.624e-6m。kd越小表明亲和力越强,进而实时观察了两种蛋白之间结合的过程,所得参数将作为性能比较的重要依据。
本发明提供了一种人源rank胞外段蛋白的表达方法,包括:rank胞外段蛋白的cdna序列的全基因合成;将所述rank胞外段蛋白的密码子进行优化,并且将优化后的基因序列构建到ppic9k上,形成ppic9k-rank-n(连接了rank胞外段蛋白的目的基因的p-pic9k),其中,所述优化后的基因序列为:
cgcctcgagaaaagacagattgctcctccatgtaccagtgagaagcattatgagcatctgggaagatgctgtaacaaatgtgaaccaggaaagtacatgtcttctaaatgcactactacctctgacagtgtatgtctgccctgtggccctgatgaatacttggattcatggaatgaagaagataaatgcttgctacataaagtttgtgatacaggcaaggccttggtggccgtggtcgccggtaactcaacgacccccaggcgttgcgcttgcacggctgggtaccactggtcacaggactgcgagtgctgtcgtcgaaacactgagtgtgctcctggtttaggtgcacaacaccctttgcaactaaacaaggacacagtttgtaaaccttgtcttgcaggttatttctctgatgcattttcctccactgacaaatgtagaccctggactaattgtacttttcttggaaagagagtcgaacatcacggtacagagaagtccgatgctgtttgtagttcttccttaccagctagaaagccaccaaatgaacctcacgtttatttgccataagcggccgcattattaa;将所述ppic9k-rank-n电转化至巴斯德毕赤酵母gs115进行蛋白表达。
在上述表达方法中,其中,所述优化后的基因序列是全基因合成的。
本发明还提供了一种人源rank胞外段蛋白的纯化方法,包括:对包含所述人源rank胞外段蛋白的发酵液上清进行超滤,并用截留3kd的膜得到超滤浓缩液;对所述超滤浓缩液进行sephadexg-50凝胶过滤层析,紫外280nm监测收集各个吸收峰的蛋白样品;对所述蛋白样品进行电泳,确定并收集含有目的条带的蛋白样品;对所述含有目的条带的蛋白样品进行q-sepharose-ff离子交换层析,紫外280nm监测收集各个吸收峰的蛋白组分;对所述蛋白组分进行免疫蛋白印迹,确定含有目的蛋白的蛋白组分,并且收集所述含有目的蛋白的蛋白组分。
在上述纯化方法中,其中,通过millipore超滤系统进行所述超滤。
在上述纯化方法中,其中,在对所述超滤浓缩液进行sephadexg-50凝胶过滤层析之前,对sephadexg-50层析柱进行清洗和平衡。
在上述纯化方法中,还包括:用bca法测定所述含有目的蛋白的蛋白组分的蛋白浓度。
在上述纯化方法中,其中,在对所述含有目的条带的蛋白样品进行q-sepharose-ff离子交换层析之前,对q-sepharose-ff层析柱进行清洗和平衡。
在上述纯化方法中,其中,用0.1m的naoh清洗所述sephadexg-50层析柱和所述q-sepharose-ff层析柱。
在上述纯化方法中,其中,用ph为9.0的tris-hcl平衡所述sephadexg-50层析柱和所述q-sepharose-ff层析柱。
本发明还提供了人源rank胞外段蛋白在制备治疗骨相关疾病和结直肠癌的药物中的应用。
本发明对rank胞外段蛋白全序列进行了优化,成功表达了rank-n蛋白,并且表达量高,且表达出的蛋白具有相关的蛋白活性。
附图说明
图1示出了rank信号通路。
图1a至图1d示出了rank胞外端基因克隆和鉴定:(图1a)人rank胞外段基因生工合成后序列琼脂糖凝胶检测:从左到右侧条带依次为rank-n基因条带,ppic9k基因条带、dnamarker:dl2000,每孔上样均为5μl;(图1b)ppic9k-rank-n构建后经xhoi和noti双酶切鉴定;(图1c)rank-n构建到ppic9k后上测序比对:已经成功克隆的ppic9k-rank-n使用通用引物进行测序,其测序结果中的552bp与genebank序列一致;(图1d)ppic9k-rank-n的构建表达图。
图2a至图2f示出了酵母菌株表达rank-n和westernblot鉴定:(图2a、图2b)酵母菌株甲醇诱导表达sds-page电泳后考马斯亮蓝染色结果,泳道1为蛋白分子量marker,左侧为分子量大小对照,0,12……96小时分别为不同时间样品,单位kd,样品20μl/孔,sds-page浓缩胶5%,分离胶12%;图2a从左到右分别为每24小时加1%甲醇、每12小时加0.5%甲醇,图2b从左到右分别为每12小时加1%甲醇每12小时加1.5%甲醇。最终选定1%/12小时的甲醇补加量;(图2c-2e):(图2c)0号菌株的甲醇诱导表达,(图2d)7号菌株的甲醇诱导表达,(图2e)将在2mg/ml(0号),4mg/ml(1-6号),6mg/ml(7-8号)g418平板上生长菌株分别进行诱导96小时;取菌液上清考染后发现1-8中蛋白表达量没有明显差异;(图2f)各个时间点样品的westernblot检测,其中,至少进行三次实验重复。
图3a至图3g显示蛋白摇瓶表达、纯化和糖基化分析:(图3a)巴斯德毕赤醉母转化子甲醇利用表型md板,mm板鉴定;(图3b)饱和度30%~60%硫酸铵沉淀的上清液和沉淀:左边泳道为蛋白分子量marker,泳道1-9分别依次为饱和度,30%,40%,50%沉淀后的上清,30%,40%,50%硫酸铵沉淀后的沉淀,盐析前诱导96h的菌液上清,60%,70%硫酸铵沉淀后的沉淀;(图3c)凝胶过滤层析图,y轴为od280nm吸收值,x轴为流过层析柱的体积,蓝色线(左边峰更高的线)为样品od280nm吸收值值,红色为od254nm吸收值;(图3d)离子交换层析图,xy轴与图3e相同,垂直线为标记,标记线后开始开始0-1.0mnacl线性洗脱;(图3e)sds-page考马斯亮蓝染色图,各泳道名称见上面标注,m表示蛋白分子量marker,1-5分别为96小时菌液上清、超滤滤出液、超滤浓缩液、分子筛样品、阴离子交换柱流穿液,6-9分别表示第一个吸收峰收集的四管,左侧为分子量大小,单位kd;(图3f)bca的蛋白质定量标准曲线,图3c(rank-n)为644μg/ml;(图3g)糖肽酶f(pngasef)处理样品后sds-page考马斯亮蓝染色结果,m表示蛋白分子量marker,con为对照组,pngasef为糖肽酶f处理组,pngasef组中35kda处为糖肽酶f。sds-page中样品上样量为蛋白分子量marker5μl/孔,样品20μl/孔,sds-page浓缩胶5%,分离胶12%。
图4显示示出了rank-n可以阻断rank-rankl信号通路:不同浓度的rank-n可以阻断rank-l激活的非经典nf-κb信号通路,1、10分别表示1、10uμg/ml,rank-l为1μg/ml。
图5显示鼠源骨髓单核巨噬细胞分化破骨细胞阻断实验:鼠源骨髓单核巨噬细胞经rankl诱导5天后trap染色观察,红色箭头示破骨细胞(倒置相差显微镜×40)。从左向右,第一张图为阴性对照,没有加入rankl,鼠源骨髓单核巨噬细胞没有分化为破骨细胞,第二张图为1μg/mlrankl蛋白,鼠源骨髓单核巨噬细胞分化为破骨细胞,图片中的紫红色部分均为破骨细胞,第三张图中加入rankl及1μg/mlrank-n,结果显示加入1μg/mlrank-n即可明显阻止rankl引起的巨噬细胞的分化。
图6a显示rank-n与人rankl的结合解离曲线:将rankl与芯片耦合,通过加不同浓度的rank-n蛋白(31.25nm、62.5nm、125nm、250nm、500nm、1000nm)去结合有rankl的芯片,然后减去背景值,可得到两者的亲和解离曲线,rankon为rank与rankl结合变化,rankoff为两者解离变化。
图6b显示通过t100软件进行拟合曲线算出平衡解离常数kd:2.62e-6m。
图7a至图7d示出了rank-n抑制结直肠癌组织块体内生长:(图7a)病人来源的结直肠癌组织移植到裸鼠皮下,经过5次传代稳定。取一只裸鼠体内结直肠癌组织块移植到8只裸鼠皮下,1周后将带瘤小鼠分为两组,一组腹腔注射pbs,一组注射rank-n,2个月肿瘤块如图;(图7b)肿瘤重量比较;(图7c)肿瘤体积大小比较(*p<0.05,**p<0.01);(图7d)wb检测结果。
图8a为病人来源的dlbcl细胞(pdc)体外不加入和加入骨髓来源的基质细胞(bmsc)以及加入和不加入rank-n后培养,进行流式检测dlbcl细胞死亡(7aad+annexinv双染色)的结果。
图8b是图8a的量化结果。
本发明使用的酵母为巴斯德毕赤酵母(pichiapastoris),保藏于中国微生物菌种保藏管理委员会普通微生物中心(cgmcc,北京市朝阳区北辰西路1号院3号),编号为cgmccno.12500,于2016年05月26日保藏。
具体实施方式
下面的实施例可以使本领域技术人员更全面地理解本发明,但不以任何方式限制本发明。
本发明中使用的缩略词如下所示:
2.2.1材料和方法
2.2.1.1实验材料:巴斯德毕赤酵母gs115(复旦大学分子医学教育部重点实验室,也可从thermofisherscientific公司购买得到),大肠杆菌dh-5α菌株(tiangen公司)。
2.2.1.2实验仪器设备
2.2.1.3主要试剂
2.2.1.4相关载体
ppic9k载体(复旦大学于敏实验室馈赠)。
2.2.1.5试剂盒
琼脂糖凝胶dna回收试剂盒(axygen)、pcr产物纯化回收试剂盒(axygen)、普通质粒小提试剂盒(axygen)、pcr产物纯化回收试剂盒(transgenebiotech)、质粒大量抽提试剂盒(macherey-nagel)、kod-plus(toyobo)、甘油检测试剂盒。
2.2.1.6分子量标记
dna分子量标记:dl2000(takara)trans2kplus(transgenebiotech)
蛋白分子量标记:pagerulerplusprestainedproteinladder(26617)
2.2.1.7主要溶液配制
10×d(20%葡萄糖):溶解200gd-葡萄糖于l000ml水中,121℃高压灭菌20分钟。
生物素(500×):即0.02%生物素,称取0.01g生物素加入50ml去离子水中0.22μm滤器过滤除菌。
10×ynb(有或无氨基酸):溶解134gynb于l000ml去离子水中,磁力搅拌溶解,0.22μm滤器过滤除菌。存于4℃保存使用。
ypd培养基:溶解10g酵母提取物,20g胰蛋白胨于去离子水中定容至900ml(如制ypd平板加入15g琼脂粉),121℃高压灭菌15分钟,冷却后加入20%葡萄糖溶液100ml。
md平板:琼脂1.5g去离子水定容至100ml,121℃高压灭菌15分钟后,待温度降至50℃左右加入10ml10×ynb,200μl生物素(500×),10ml20%葡萄糖,混匀倒平板。
mm平板:琼脂1.5g去离子水定容至100ml,121℃高压灭菌15分钟后,待温度降至50℃左右加入10ml10×ynb溶液,生物素(500×)200μl,10%(v/v)甲醇10ml,混匀倒平板。
bmgy培养基(1l):10g酵母提取物,20g胰蛋白胨,溶于700ml去离子水中,121℃高压灭菌15分钟,冷却至室温,加入1m磷酸钾缓冲液(ph6.0)100ml,10×ynb溶液100ml,生物素(500×)2ml,10%(v/v)甘油100ml。
bmmy培养基:10g酵母提取物,20g胰蛋白胨,溶于700ml去离子水中,121℃高压灭菌15分钟,冷却至室温,加入1m磷酸钾缓冲液(ph6.0)100ml,10×ynb溶液100ml,生物素(500×)2ml,在随后使用时加入甲醇。(根据实际甲醇终浓度)
发酵无机盐培养基(bsm):sodiumcitrate·2h20-1.5g,caso4·2h20-1.01g
k2so4-18g,mgso4-7.32g,koh-4.13g,85%h3po4-27ml,甘油-32ml,补去离子水至1l,121℃1.5kg/cm2高压灭菌20分钟
微量元素ptm1:cuso4·5h2o-6g,mnso4·h2o-3g,h3bo4-0.02g,zncl2-20g
ki-0.8g,namoo4·2h2o-0.2g,cocl2·6h2o-0.49g,feso4·7h2o-65.06g,h2so4-10ml,caso4·2h2o-0.5g,mgso4-1.71g,生物素-0.2g,0.22μm滤器过滤除菌,存于4℃保存使用,用量为每1lbsm中加2mlptm1。
pbs:在800ml蒸馏水中溶解nacl-8g,kcl-0.2g,na2hpo4·12h2o-3.6g
kh2po4-0.24g,定容至1l,121℃高压灭菌20分钟后,室温保存。
10xruningbuffer(1l):trisbase-30.2g,glycine-188g,溶于1l去离子水中
5xloadingbuffer(裂解蛋白):1mtris-hcl(ph6.8)-12.5ml,sds-5g,溴酚蓝-0.25g,甘油-25ml,dtt-3.855g。
50×平衡缓冲液tris-hcl(ph9.0)1l:取121.14gtris,加入800ml去离子水溶解,室温下用浓盐酸调ph至9.0,加水定容到1l。纯化中使用浓度为0.02m。
洗脱缓冲液:取58.5gnacl,用1x平衡缓冲液tris-hcl(ph9.0)溶解。
考马斯亮蓝染液1l:r-250-2.5g,95%乙醇-500ml,蒸馏水-450ml,冰醋酸-50ml。
脱色液1l:乙醇-500ml,冰醋酸-100ml,蒸馏水400ml。
sds-page配方
浓缩胶:
浓缩胶(5%acrylamide)
分离胶:
(4)12%gel
2.2方法
2.2.1.目标基因全基因合成:
从genebank基因库中搜索目标蛋白rank胞外段的cdna序列,并对cdna序列进行巴斯德毕赤酵母表达系统的密码子优化,送于上海生工生物工程股份有限公司进行rank胞外段的基因序列合成。
2.2.2rank-n密码子优化,并构建到p-pic9k上
2.2.2.1rank-n巴斯德毕赤酵母密码子优化
优化前:
cgcctcgagaaaagacagatcgctcctccatgtaccagtgagaagcattatgagcatctgggacggtgctgtaacaaatgtgaaccaggaaagtacatgtcttctaaatgcactactacctctgacagtgtatgtctgccctgtggcccggatgaatacttggatagctggaatgaagaagataaatgcttgctgcataaagtttgtgatacaggcaaggccctggtggccgtggtcgccggcaacagcacgaccccccggcgctgcgcgtgcacggctgggtaccactggagccaggactgcgagtgctgccgccgcaacaccgagtgcgcgccgggcctgggcgcccagcacccgttgcagctcaacaaggacacagtgtgcaaaccttgccttgcaggctacttctctgatgccttttcctccacggacaaatgcagaccctggaccaactgtaccttccttggaaagagagtagaacatcatgggacagagaaatccgatgcggtttgcagttcttctctgccagctagaaaaccaccaaatgaaccccatgtttacttgccctaagcggccgcattattaa
优化后:
cgcctcgagaaaagacagattgctcctccatgtaccagtgagaagcattatgagcatctgggaagatgctgtaacaaatgtgaaccaggaaagtacatgtcttctaaatgcactactacctctgacagtgtatgtctgccctgtggccctgatgaatacttggattcatggaatgaagaagataaatgcttgctacataaagtttgtgatacaggcaaggccttggtggccgtggtcgccggtaactcaacgacccccaggcgttgcgcttgcacggctgggtaccactggtcacaggactgcgagtgctgtcgtcgaaacactgagtgtgctcctggtttaggtgcacaacaccctttgcaactaaacaaggacacagtttgtaaaccttgtcttgcaggttatttctctgatgcattttcctccactgacaaatgtagaccctggactaattgtacttttcttggaaagagagtcgaacatcacggtacagagaagtccgatgctgtttgtagttcttccttaccagctagaaagccaccaaatgaacctcacgtttatttgccataagcggccgcattattaa
优化后的序列送公司(生工)全基因合成。
2.2.2.2ppic9k-rank-n大量抽提(mn
(1)过夜摇菌,将ppic9k-rank-n阳性克隆菌株菌液10μl加入200mllb培养基(加200μl氨苄青霉素)中,250rpm37℃过夜培养,该菌株需培养22小时左右。
(2)离心管收集菌体,用4个50ml离心管收集菌液离心4000rpm,15分钟,弃上清,尽量弃净。
(3)重悬,用bufferres10ml震荡重悬所有菌体。
(4)细胞裂解,加入10mlbufferlys(确保用之前sds没有析出),轻轻上下颠倒5次,室温静置5分钟。
(5)平衡柱子,在柱子上沿璧旋转加入12mlbufferequ,让其重力作用自然流出。
(6)中和,4中液体中加入10mlbufferneu,上下轻轻颠倒离心管10到15次。
(7)混匀和上柱,将离心管中液体混匀后直接倒入平衡好的柱子上,让液体随重力自然流出。
(8)洗涤,待柱子快流尽时,旋转加入10mlbufferequ洗一遍。
(9)去掉柱子里的滤纸。
(10)洗涤,柱子里居中垂直加入16mlwashbuffer让液体随重力自然流出。
(11)洗脱,待其快流尽,加入5mlelutionbuffer(使用前在50℃水中孵育10分钟),用15ml离心管收集洗脱液。
(12)沉淀,洗脱液中加入3.5ml异丙醇,混匀。
(13)分装到6个ep管中,离心12000rpm4℃30分钟。
(14)弃上清,每个ep管中加入70%乙醇再洗涤一遍离心15000g5分钟4℃,弃上清,室温静置10分钟。
(15)每管加20-30μl去离子水震荡混匀后合并在一起,用nanodrop测定浓度,-30℃保存。
2.2.2.3将合成的序列、载体ppic9k用xhoi和noti双酶切
反应体系40μl:
37℃水浴箱1小时到2小时。
2.2.2.4双酶切产物切胶回收,连接,转化,摇菌,小抽鉴定
双酶切产物切胶回收
将酶切产物进行凝胶电泳(六孔梳1%琼脂糖30分钟),用手术刀片切取含目的片段的琼脂糖凝胶块,rank-n(550bp左右)、ppic9k(9000bp左右),凝胶回收试剂盒(axygen公司)回收获取目的条带,步骤如下:
(1)切胶样品用天平称重后加3倍体积的buffera。
(2)将样品放在75℃水浴锅溶解,6到8分钟,期间间隔2分钟,颠倒ep管。
(3)加入0.5倍体积buffera的bufferb。
(4)柱子放到收集管上,将样品冷却后加入柱子,室温放置1分钟。
(5)离心12000rpm1分钟,弃收集管中废液。
(6)沿柱子内壁加入500μlbufferw1。
(7)离心12000rpm1分钟,弃收集管中废液,重复步骤4,5。
(8)沿柱子内壁加入700μlbufferw2,离心12000rpm1分钟,弃收集管中废液,重复步骤8。
(9)将柱子放在空的收集管中,离心12000rpm2分钟。
(10)取一个新的ep管,将柱子放在上面,室温静置1分钟。
(11)柱子上加入25μl去离子水,室温静置1分钟离心12000rpm1分钟。
(12)此时ep管中即为纯化的dna,用nanodrop测定浓度后,-30℃保存待用。
2.2.2.5胶回收连接
连接反应体系(10μl):
16℃连接过夜。
2.2.2.6连接产物转化e.colidh-5α
(1)从-80℃冰箱中取出dh5α感受态,冰上融化。
(2)5μl连接产物放入50μl的dh5α中,冰上放置30分钟。
(3)42℃水浴箱孵育90秒。
(4)迅速冰浴5分钟。
(5)超净台内加入800μl无抗生素的lb培养基,37℃180rpm1小时。
(6)离心5000rpm3-4分钟,吸去上清800μl。
(8)将剩余200μl菌液吹匀涂布在含氨苄抗生素的lb平板上。37℃倒置过夜培养。
(9)14-18小时后检查是否有单克隆菌落长出。
2.2.2.7质粒抽提
质粒抽提试剂盒(axygen公司)过程如下:
(1)挑单克隆过夜摇菌:将lb板上长的菌株挑取到4ml带氨苄抗生素培养基中(15ml无菌离心管或者试管),250rpm37℃过夜培养,一般12-16小时。
(2)用ep管收集菌液:在超净台中将离心管中菌液倒入ep管中(一般一次只能倒入1.5ml),离心12000rpm1分钟,弃上清。再在相同的ep中倒入同编号菌液1.5ml,离心12000rpm1分钟,弃上清。
(3)加入250μlbuffers1,震荡混匀。
(4)加入250μlbuffers2,轻轻颠倒混匀5-10下,达到澄清。
(5)加入350μlbuffers3,颠倒混匀5-10下,可出现白色絮状物质。
(6)离心12000rpm10分钟。
(7)柱子放到收集管上,小心吸取上清加入柱子,室温放置1分钟。
(8)离心12000rpm1分钟,弃收集管中废液。
(9)沿柱子内壁加入500μlbufferw1,离心12000rpm1分钟,弃收集管中废液。
(10)沿柱子内壁加入700μlbufferw2,离心12000rpm1分钟,弃收集管中废液,重复步骤10。
(11)将柱子放在空的收集管中,离心12000rpm2分钟。
(12)取一个新的ep管,将柱子放在上面,室温静置1分钟。
(13)柱子上加入60μl去离子水,室温静置1分钟,离心12000rpm1分钟。
(14)此时ep管中即为纯化的dna,用nanodrop测定浓度后,-30℃保存待用。
2.2.2.8抽提质粒用xhoi和noti进行双酶切鉴定
反应体系10μl:
37℃水浴1小时到2小时后,将酶切产物进行凝胶电泳(15孔梳1%琼脂糖30分钟)出现9k+0.5k条带即为阳性克隆,取条带匹配样本2-3个送公司测序(由上海桑尼生物公司完成),同时取阳性克隆菌株菌液等体积加入50%无菌甘油-80℃保存。
2.2.3.ppic9k-rank-n电转化巴斯德毕赤酵母gs115并筛选获得稳转株
2.2.3.1取测序正确的ppic9k-rank-n大量抽提(mn
2.2.3.2ppic9k-rank-n线性化
单酶切反应体系100μl:
37℃水浴过夜酶切,酶切的质粒用pcr回收试剂盒(transgenebiotech)回收,步骤如下:
(1)样品加5倍体积的溶液bb,震荡混匀。
(2)柱子放到收集管上,将样品加入柱子,室温放置1分钟。
(3)离心12000rpm1分钟,弃收集管中废液。
(4)沿柱子内壁加入650μlwb。
(5)离心12000rpm1分钟,弃收集管中废液,重复步骤4,5。
(6)将柱子放在空的收集管中,离心12000rpm2分钟。
(7)取一个新的ep管,将柱子放在上面,室温静置1分钟。
(8)柱子上加入30-50μl去离子水(提前65℃水浴10分钟),室温静置1分钟,离心12000rpm1分钟。(可再重复一次)
(9)用nanodrop测定浓度后,-30℃保存待用。
2.2.3.3巴斯德毕赤酵母gs115菌种感受态制备
(1)挑gs115单克隆菌落于5mlypd培养基,30℃,250rpm,培养12-15小时。
(2)取培养液200μl,接于200mlypd培养基的1l摇瓶中,30℃,250rpm,培养至od600=0.6-0.8。
(3)冰浴15分钟,4℃,无菌条件下转至50ml离心管中,3000rpm,离心5分钟。
(4)弃上清,无菌条件下加入50ml预冷的无菌水重悬菌体,冰浴15分钟,4℃,3000rpm,离心5分钟。
(5)重复步骤4。
(6)弃上清,无菌条件下加入40ml预冷的山梨醇重悬菌体,冰浴15分钟,4℃,
3000rpm离心5分钟。
(7)重复步骤6。
(8)弃上清,无菌条件下加入1ml预冷的山梨醇重悬菌体,分装,120μl/支,冰浴备用。
2.2.3.4ppic9k-rank-n电转化巴斯德毕赤酵母gs115:
(1)取线性化的ppic9k-rank-n质粒30μg,加入120μlgs115菌种感受态。
(2)混匀后转入无菌已遇冷的电转化杯中。
(3)电转仪设置成真菌模式,将电转化杯放入电转仪固定铁片间,按pμlse键进行电转。
(4)待脉冲声音响过之后,立即加入1ml预冷1m山梨醇。
(5)30℃培养1小时,分500μl涂布于15cmmd板上,48小时后观察平板上是否有单克隆菌落生长。
2.2.3.5筛选多拷贝酵母转化子
(1)72小时后his-md板上长出his+转化子,用灭菌牙签点取单克隆菌株4x96个,于96孔板中30℃培养(含ypd200μl/孔)。
(2)1天后将96孔板中菌取10μl转移到一个新的96孔板中30℃培养(含ypd200μl/孔)。
(3)1天后,取5μl菌液接种于2mg/mlypd-g418(遗传霉素)平板上,30℃培养3天后选取长出菌株依次接种于4mg/ml板子上进行,重复上述过程取长出菌株依次接种6mg/ml、8mg/ml.......,g418可以筛选到直到没有菌生长为止(必须保证g418的抗菌活性)。保留仅在各浓度梯度下生长菌株2-3株。本次试验于6mg/mlypd-g418平板上筛选出两株多拷贝阳性酵母转化菌株。
2.2.3.6巴斯德毕赤酵母转化子甲醇利用表型md板、mm板鉴定
(1)准备md,mm平板。
(2)将待鉴定的菌落在mm、md板上四区划线并做好标记。
(3)30℃培养3天待克隆长出后比较菌落大小。md板大,mm板小,为甲醇利用慢型,md板和mm板上菌落一样大,为甲醇利用快型。
2.2.3.7菌株甲醇诱导表达
(1)将2mg/ml、4mg/ml、6mg/mlypd-g418平板上筛选的菌株四区划线于ypd平板上,待出现单克隆菌落。
(2)挑单克隆菌落接种于5mlbmgy培养基中,30℃250rpm过夜培养。
(3)菌液1:100接种到100mlbmgy中,30℃250rpm培养,检测od600数值。
(4)至对数生长期(od达到2-6之间,约17小时)时,留样200μl,留样样品离心4000rpm,5分钟,取上清160μl,-20度存放。同时换bmmy培养基并加入甲醇至终浓度1%开始诱导。(上清若浑浊,多为大肠杆菌污染)。
(5)每12小时后补加100%甲醇至终浓度1%开始诱导表达,留样200μl,留样样品离心4000rpm,5分钟,取上清160μl,-20度存放。
(6)诱导96小时后,收集摇瓶培养液,4℃离心4000rpm,20分钟,收集上清备用。
(7)检测表达将0-96h样品加5xloading40μl,100℃煮沸10分钟,12%分离胶sds-page,150v,约65分钟。加考马斯亮蓝染色液于摇床上过夜染色,脱色处理后观察结果。
2.2.3.7gs115菌冻存
(1)将gs115菌株四区划线于ypd平板。
(2)2至3天后,挑单克隆菌株于5mlypd培养基中30℃250rpm过夜培养。
(3)将过夜培养菌按1:100接种到含100mlypd的摇瓶中30℃250rpm培养。
(4)待od600长到5左右,3000rpm4℃离心5分钟收集菌体。
(5)用含50%甘油ypd培养基10ml重悬菌体,每ep管100μl冻存于-80℃。
2.2.4免疫蛋白印迹westernblot
将蛋白样品加入5×loadingbuffer100℃加热10分钟变性,进行westernblot检测或储存于-20℃。
(1)上样:将20ug-40ug蛋白样品加入配好的12%sds-page胶中;
(2)电泳:浓缩胶恒压80v40分钟,分离胶恒压120v60分钟;
(3)转膜:将pvdf膜(4.5cmx8cm)泡在甲醇中5分钟,转膜液冰上预冷30分钟;采用夹心法转膜,顺序为:黑色塑料板(负极)-海绵-滤纸-胶-pvdf膜-滤纸-海绵-白色塑料板(正极),再将转膜夹按照正确电极方向放入转膜槽中,加入转膜液,冰块,恒压100v冰浴中转1小时;
(4)封闭:5%脱脂牛奶放在摇床上1-2个小时。
(5)一抗:按适宜一抗浓度稀释抗体(按抗体说明书上要求),4℃孵育过夜。
(6)洗膜:pbs-0.05%tween20洗膜三次,每次8分钟。
(7)二抗:加入适当浓度的hrp标记二抗(按抗体说明书上要求),室温孵育1-2小时。
(8)洗膜:pbs-0.05%tween20洗膜三次,每次5分钟。
(9)显影:用piercewestpico底物或者millipore显影,fujifilmlas4000扫膜成像。
2.2.5ppic9k-rank-n菌株高密度发酵
接种:
(1)-80℃低温冰箱中取出菌株,划ypd平板。
(2)2至3天后,待平板长出单克隆,挑单克隆菌株于5mlypd培养基中30℃250rpm过夜培养,此为一级种子液。
(3)将一级种子液1:500放大到200mlypd摇瓶中,30℃250rpm培养约12小时,达到od6006-8,作为二级种子液。
发酵:
(1)在5l发酵罐中装入3lbsm无机盐培养基,121℃高压灭菌20分钟,冷却至室温,加入ptm16ml,用氨水调节ph至5.0。
(2)接入二级种子液200ml,溶氧(do)控制为35%,搅拌速度为500rpm,温度设置为30℃,开始发酵,每隔4小时测定一次od600。od600超过100后搅拌速度改为650rpm。待发酵罐中甘油被耗尽后,加入甲醇开始诱导,添加甲醇量在4小时内从0.4ml/分钟到4ml/分钟,以后一直维持此速度。每隔4小时测定发酵液od,并离心收上清液。诱导24小时后进行还原型sds-page蛋白电泳、考染检测以确定是否有目的蛋白表达。诱导36小时后下罐,收集发酵液上清4℃保存,随后进行蛋白质的纯化工作。
2.2.6蛋白纯化
2.2.6.1硫酸铵分级沉淀
首先配制1l硫酸铵饱和溶液。取50ml酵母菌上清液于500ml离心管中,以此为基准体积计算不同饱和度硫酸铵溶液的加入体积(参照硫酸铵沉淀法纯化蛋白配比用表)。实验硫酸铵饱和度每10%为一间隔,范围从30%到70%。实验在4℃条件下进行,用1cm搅拌子搅拌,加完硫酸铵后持续搅拌4小时,再置离心管于4℃高速离心机上以每分钟10000转离心40分钟。取上层清液样于一干净容器内,下层沉淀用q-sepharose-ff阴离子交换柱平衡缓冲液重悬-20℃保存,备还原型sds-page蛋白电泳、考染鉴定。接下来以前次上层清液样体积为基数计算下一步饱和硫酸铵溶液的体积,应扣除前次实际加入体积,然后重复前面的操作至70%硫酸铵饱和度。
sds-page鉴定:10孔12%sds-page胶,每孔加20μl蛋白样品。考马斯亮蓝染色4小时,脱色2h至目的蛋白条带清晰为止。
2.2.6.2超滤—sephadexg-50—q-sepharose-ff法纯化:
(1)超滤,发酵液上清通过millipore超滤系统用截留3kd的膜浓缩到500ml。
(2)sephadexg-50凝胶过滤层析,使用前先用0.1mnaoh清洗sephadexg-50层析柱1-2柱体积(cv),接着用tris-hcl(ph9.0)平衡2个cv以上。将超滤浓缩液约500ml以5ml/分钟过柱,0.02mtris-hcl(ph9.0)缓冲液洗脱,流速8ml/分钟。紫外280nm监测收集各个吸收峰下蛋白。
(3)电泳检测目的蛋白峰,凝胶过滤层析后收集样品进行电泳,考马斯亮蓝染色脱色后,确定是否含有目的条带的组分。
(4)q-sepharose-ff离子交换层析,使用前用0.1mnaoh清洗q-sepharose-ff1个柱体积,100%b将b通道充满洗脱缓冲液(1mnacltris-hcl)过柱1个cv,0.02mph9.0的tris-hcl平衡10个柱体积后以5ml/分钟开始上样[(3)中收集的目的蛋白组分]。样品上样结束后,速度调为8ml/分钟平衡缓冲液清洗柱子4个柱体积,接下来将b通道调为20%100分钟,8ml/分钟进行连续梯度洗脱,紫外280nm监测各个蛋白吸收峰并收集各组份蛋白(留意观察,若吸收峰出现拐点应及时换新管收集蛋白),各组份蛋白使用考马斯亮蓝染色脱色和免疫蛋白印迹确定含有目的蛋白的组份。bca法测定蛋白浓度。纯化后的目的蛋白分装,冷冻抽干。
2.2.7rank-n功能验证
(1)raw264.7细胞铺12孔板,106细胞/孔,共12孔。
(2)实验设4个组,分别为阴性对照组,rankl(50ng/ml)组,rank-n(1μg/ml)、rankl(50ng/ml)组和rank-n(10μg/ml)、rankl(50ng/ml)组。每组设置3个复孔。
(3)24小时后,收样,将细胞转移到ep管中,500g4分钟离心去上清。
(8)用1mlpbs重悬细胞,500g,4分钟离心去上清。
(9)用80μl1xloading重悬细胞,100℃10分钟,样品冻存于-30℃冰箱。
(10)westernblot检测,方法同2.2.4。
2.2.8阻断rank介导的非经典nf-κb信号通路
2.2.8.1rank-n阻断鼠源骨髓单核巨噬细胞分化实验
小鼠bmm(骨髓单核巨噬细胞)提取及诱导成破骨细胞:
准备器材及试剂:75%乙醇,1xpbs(无菌),1ml注射器,剪刀、镊子、灭菌草纸等手术器械,dmem培养液、fbs、双抗,红细胞裂解液(rbclysisbuffer):nh4cl4.1425g,khco30.5006g,edta-na20.018612g,500mlddh2o配置,调ph至7.2-7.4,m-csf(r&d),rankl(r&d)。
实验动物:
5周龄c57的雄性小鼠,体重25-30g,购于上海上海斯莱克实验动物有限公司,喂养于上海中科院健康所动物实验中心,环境温度保持在22±2℃,并保证12小时昼夜节律,普通鼠食喂养。
破骨细胞的诱导及培养:
1.取5周龄c57小鼠4只,腹腔注射1.2%的三溴乙醇溶液,小鼠的剂量为0.2ml/10克。麻醉后处死,置于75%的酒精中浸泡10分钟。
2.剪断股骨和胫骨两端,在6cm培养皿中用1ml注射器吸取加1%双抗和10%fbs的dmem冲洗骨头至泛白,移液器将细胞团吹散,70μm尼龙膜过滤去大块,1000rpm离心5分钟。
3.去上清,红细胞裂解液裂红后,1000rpm离心5分钟后,去上清,用含1%双抗10%fbs的dmem重悬细胞。(可以忽略不做)
4.将细胞接种于10cm培养皿中,加入含1%双抗10%fbs的dmem,并加入m-csf(终浓度为20ng/ml,r&d),37℃,5%co2的培养箱培养2天,贴壁细胞视为bmm。
5.胰酶消化掉贴壁细胞30分钟,然后细胞刮刮下细胞并计数,1x105细胞/孔l接种于6孔板中诱导成破骨细胞,加入含1%双抗10%fbs的dmem,并加入m-csf(终浓度为20ng/ml),贴壁2天,换液,加入m-csf,rankl(终浓度为50-100ng/ml)37℃,5%co2的培养箱培养,至第5-7天检测破骨细胞形成情况,为后续做破骨细胞分化检测试验做准备。(24孔板,5x103细胞/孔,做破骨细胞分化检测)。
2.2.8.2破骨细胞(trap)染色观察:
取3ml鼠源骨髓单核巨噬细胞的于24孔板中,每孔1ml,细胞数约为5x103细胞/孔,共3孔。待细胞贴壁后,将培养细胞分为3组,其中一孔加入1μg/mlrankl蛋白,一孔同时加入1μg/mlrank-n,另外一孔做阴性对照。37℃,5%co2的培养箱培养,至第5-7天检测破骨细胞形成情况,用细胞抗酒石酸酸性磷酸酶(trap)活性染色试剂盒染色,再用倒置相差显微镜观察拍照。
2.2.9biacoret100实验检测rankl和rank的亲和平衡常数
bia技术是基于一种表面等离子共振(spr)的物理光学现象的生物传感分析技术。不必使用荧光标记和同位素标记,从而保持了生物分子的天然活性。传感器芯片是实时信号传导的载体。本次试验我们选用了用途最广的cm5芯片,传感片cm5表面覆盖了一层葡聚糖。带有化学基团如-nh2,-sh,-cho,-oh,-cooh的生物分子(ligand)可通过化学反应,以共价键耦联的方式与葡聚糖上的羧基耦联。从而使生物分子耦联到传感片表面。
1、缓冲液准备:对1xpbs缓冲液进行高压灭菌和超声脱气处理。
2、预结合实验:将样品溶解在不同ph值的缓冲液中,使其带上不同量的电荷。然后,将样品流过芯片表面,观察它与芯片的结合曲线,判断出合适的条件。
3、测试:最终选定用ph5.5的醋酸钠低速流过cm5,使芯片与葡聚糖上的羧基结合。
4、用100μlrankl蛋白溶液(280nm)通过cm5芯片表面,与芯片耦合。
5、用100μlrank蛋白溶液(1000nm)上样,使其通过结合有rankl的芯片,并用buffer解离5分钟。
6、将rank浓度用1xpbs缓冲液稀释成合适的浓度依次为1000nm、500nm、250nm、125nm、62.5nm、31.25nm,使其通过结合有rankl的芯片并解离。
6、通过t100中分析软件得到rank与rankl的亲和解离常数。
2.2.10病人来源的异种移植(patientderivedxenograft,pdx)模型
裸小鼠:babl/c遗传背景,t细胞免疫缺陷小鼠,购买于上海斯莱克实验动物中心,饲养于中科院上海生命科学研究院健康科学研究所spf级动物房;
使用人结直肠癌标本建立裸鼠肿瘤模型:从上海中山医院采集新鲜肿瘤标本,将新鲜的肿瘤标本先用生理盐水反复冲洗后剪去非肿瘤组织及坏死部分,然后浸泡于加入双抗的dmem培养基的无菌离心管中,放在冰盒里返回实验室。
皮下移植:在无菌超净台中取出离心管倒入平皿中,先用三溴乙醇腹腔麻醉小鼠(4周龄),后用眼科剪将组织剪成3*3*3mm3的小块。小鼠取左或右侧卧位,常规消毒后分别在左右腋中线处纵向剪一小切口,用弯眼科镊从切口处在皮下(不要戳破)钝性分离出适度腔隙,用直眼科镊将结直肠癌组织块塞入腔隙内,皮钉固定切口。(若塞入两个以上肿瘤块应让其在腔隙内排成条状便于血供)
肾囊膜移植:在培养皿中,用眼科剪将组织剪成1*1*1mm3小块,然后三溴乙醇腹腔麻醉小鼠(4周龄)。小鼠取右侧卧位,常规消毒后从左侧肾脏部位垂直脊椎切开一约8mm的皮肤切口后平行切开7mm的肌肉切口,打开腹腔托出肾脏。用血管钳将6块淋巴瘤组织小块(约1*1*1mm3)接种于肾囊膜下,还纳肾脏于腹腔,分层缝合肌层及皮肤层。
待第一代荷瘤鼠瘤块长至800mm3时(约2个月),取出瘤块长至800mm3一部分进行传代接种;一部分标本浸泡于4%的多聚甲醛中固定送于中山医院进行病理学检测;另一部分瘤块修剪成体积2*2*2mm3大小放入装有冻存液的速冻管中于超低温冰箱中(-80℃)过夜,次日取出放入-196℃的液氮罐中,以备日后复苏使用。如此连续传代至第五代,观察每一代的瘤体生长情况及移植瘤组织病理学变化。五代后肿瘤组织性状稳定,后续rank-n的成药性实验均使用五代后荷瘤裸鼠作为实验鼠。将稳定的结直肠癌肿瘤组织块(3*1*1mm3)移植到裸鼠肾囊膜或皮下一周后,腹腔注射rank-n(10mg/kg)或者等体积pbs,两天一次,1-2个月后取出肿瘤块分析。
2.2.11病人来源的弥漫型大b细胞淋巴瘤细胞(dlbclpdc)和小鼠骨髓基质细胞(bmsc)共培养
首先将c57野生型小鼠体内骨髓细胞取出铺到24孔板中(约5到6只一个24孔板),在dmem10%fbs血清中培养5天后细胞长满。接下来将裸鼠肾囊膜下dlbcl肿瘤块取出、剪碎、消化成单细胞,使用ficoll分选出单核细胞2*105/ml接种于长满bmsc中24孔中。共同培养4天后检测。
2.2.12facs染色分析
(1)细胞4℃离心500g4分钟,弃掉上清。
(2)加入1ml遇冷的pbs(含0.5%fbs)重悬细胞,500g,4分钟4℃离心,弃掉上清。
(3)用100μlpbs(含0.5%fbs)重悬细胞,加入annexinv、7-aad分别5μl。同时做只加入annexinv或者7-aad以及不加染料对照组。4℃避光染色30分钟。
(4)染色结束后加入1mlpbs(含0.5%fbs),轻轻吹打细胞,500g4分钟4℃离心,弃掉上清。
(5)重复步骤4。
(6)用300μlpbs(含0.5%fbs)重悬细胞,上机检测。
3.结果
3.1克隆获得rank胞外段基因
成熟rank的胞外蛋白片段为全长蛋白的,在pubmed-gene查出人rank全长序列选择胞外段(n端)29-213aa,相应mrna长度为552bp。对胞外段进行巴斯德毕赤酵母密码子优化后的序列送公司(生工)全基因合成。琼脂糖凝胶电泳结果显示此条带略大于500bp(见图1a)ppic9k-rank-n构建后xhoi和noti双酶切鉴定(见图1b)。酶切产物使用胶回收试剂盒回收,使用t4连接酶连接1小时,连接产物转化dh5α细菌。摇菌质粒小抽后,将获得的质粒再次用xhoi和noti双酶切鉴定,双酶切产物进行琼脂糖凝胶电泳,电泳结果呈现两条带(9000bp+500bp)为阳性克隆,鉴定为阳性克隆的质粒送公司测序。在pubmed网站上用blast进行比对,测序结果插入片段序列与genebank网站中rank胞外段序列完全一致(图1c)。
3.2筛选获得rank-n较高表达巴斯德毕赤酵母菌株
线性化ppic9k-rank-n并电转化巴斯德毕赤酵母gs115,通过组氨酸缺陷和g418遗传霉素两轮筛选,8株g418较高抗性菌株被筛选得到,在此将其编号为1-8,其中1-6在4mg/mlg418平板上生长菌株,7、8在6mg/mlg418上生长(图2e)。分别将这些菌株进行甲醇诱导表达:具体方法见方法2.2.3.5,保持甲醇诱导浓度为1%/12小时。96小时后停止诱导,将收集的样品进行sds-page电泳,考马斯亮蓝染色脱色后(具体见方法)。发现在目的蛋白大小区域(27kd)有一个明显的逐渐增加的蛋白带(图2d)。接下来用rank抗体对2号菌株诱导表达的上清进行免疫蛋白印记分析。该条带被人rank抗体特异性识别,证明该条带为rank-n蛋白(图2f)。
3.3rank-n表达菌株蛋白摇瓶表达、纯化和糖基化鉴定
在rank-n表达菌株蛋白摇瓶表达纯化工作中我们首先在实验室小体系摇瓶中进行预实验,经过摇瓶表达发酵的60%硫酸铵沉淀(3b)-3k半透膜透析-qsepharoseff离子交换层析三个步骤纯化重组蛋白。优化条件并得出有效的纯化数据。
在2.2l实验室摇瓶诱导表达96h后离心收集酵母培养上清,2l菌液上清被收集后使用截留3kd超滤膜进行超滤,收集超滤后截留的组分体积约为500ml。接下来样品用akta仪器进行sephadexg-50凝胶层析。收集od280紫外吸收峰下的样品进行聚丙烯酰胺凝胶电泳(sds-page),考马斯亮蓝染色显示含目的蛋白组分出现在第一个紫外吸收峰中(图3c)。收集到样品约650ml,这时候盐分和部分色素已经被去除。将含有目的蛋白组分用q-sepharose-ff阴离子交换柱继续纯化,0-1.0mnacl线性洗脱,目的蛋白组分在0.03molnacl通道处被收集获得(第一个紫外吸收峰1500ml处,见图3d箭头处),共收集到3管,分别45ml。分子筛收集到样品和离子交换纯化后获得的样品进行sds-page考马斯亮蓝染色后结果(见图3e)。并进行bca的蛋白质定量(图3f),梯度稀释样品,并测定od570,绘制曲线,得到方程y=1618.2x+3.6356,r2=0.9995。为了证实表达纯化的蛋白大小(约28kd)与理论大小(21kd)是不是由于糖基化导致的,我们将纯化的蛋白用去除n-糖基化的糖肽酶f(pngasef)处理,发现处理后蛋白大小变小为21kda,与理论一致(图3g),因此该蛋白发生了n-糖基化。
3.4rank-n的体外功能
3.4.1功能性rank-n可以体外阻断rank-rankl信号介导的非经典nf-κb信号通路
为了探索rank-n是否具有阻断rank介导的信号转导功能,进行如下实验。raw264.7细胞在rankl作用24小时后p100剪切成p52明显增加,在仅仅加入1μg/ml人rank-n后,p52的增加相对于对照组减少,且这种减少随着rank-n浓度增加而更加明显。而在rank-n加入后traf3的降解没有对照组明显同时relb的增加相对于对照组减少(图4)。p100剪切成p52也没有对照组多这些实验证明rank-n可以体外阻断rank介导的非经典nf-κb信号通路激活。
3.4.2人rankl蛋白促进鼠源骨髓单核巨噬细胞分化和rank-n阻断破骨细胞分化实验
取3ml鼠源骨髓单核巨噬细胞的于24孔板中3个孔,每孔1ml,细胞数约为5x103细胞/孔,其中一孔加入1μg/mlrankl蛋白,另一孔加入50ng/mlrankl及1ug/mlrank-n,还有一孔做为空白对照。培养五天后,可以观察到鼠源骨髓单核巨噬细胞分化为破骨细胞:多个单核细胞融合、体积增大的巨细胞,用trap试剂染色可清楚观察(图5)。
3.4.3biacoret100实验测定人rank和rankl蛋白之间的相互作用
我们通过biacoret100进一步验证rank和rankl的相互作用,通过软件测定平衡解离常数kd值,直观反映出两者的亲和力。本次试验选用cm5芯片,使rankl以共价键耦联的方式与葡聚糖上的羧基耦联。首先将rankl与芯片耦合,将rank浓度用1xpbs缓冲液稀释成合适的浓度依次为1000nm、500nm、250nm、125nm、62.5nm、31.25nm,使其通过结合有rankl的芯片。当两者结合时cm5芯片表面物质的质量会发生一个动态变化,这时t100会记录下对应的响应值(response)。通过软件处理数据后,计算得出两者的平衡解离常数(kd),结果如图6a和图6b所示。
3.5rank-n的体内功能
功能性rank-n可以减弱病人来源结直肠癌的体内生长,抑制弥漫型大b细胞淋巴瘤的体外生长和存活
利用已经建成的稳定的病人来源的结肠肿瘤块移植模型(pdx)系统,用rank-n进行处理(腹腔注射,两天一次,10mg/kg),2个月后观察结直肠癌的体内生长情况,发现rank-n处理组肿瘤无论是质量和体积都显著小于对照组(图7a-c)。因此rank-n是可以阻断肠癌肿瘤体内生长。
为了探索rank-n抑制结直肠癌pdx肿瘤体内生长的机制,我们检测了两组肿瘤中与肿瘤生长密切相关的分子表达情况。首先,我们检测发现p52在rank-n作用后是减少的,同时traf3的降解被抑制,共同证明了rank蛋白在体能阻断rank激活的非经典
参见图8a和图8b,示出了rank-n可以抑制弥漫型大b细胞淋巴瘤(dlbcl)肿瘤细胞存活:图8a为病人来源的dlbcl细胞(pdc)体外不加入和加入骨髓来源的基质细胞(bmsc)以及加入和不加入cd40-n后培养后,进行流式检测细胞死亡(7aad+annexinv双染色)的结果(左上角图为不加入bmsc和cd40-n,左下角图加入bmsc,右上角图加入cd40-n,右下角图加入bmsc和cd40-n);图8b是图8a的量化结果,cd40-n为1,10μg/ml。(至少三次实验重复,**p<0.01。)结果显示rank-n能抑制dlbcl肿瘤细胞的存活。
4.讨论和结论
rankl/rank/opg系统是近年来发现的在破骨细胞分化过程中具有非常重要作用的信号传导通路。成骨细胞、骨髓基质细胞及活化的t淋巴细胞表达rankl与破骨细胞表面的rank结合后促使破骨细胞分化增殖。研究发现,rankl–rank–opg通过作用于dc影响免疫,也可以通过调节破骨细胞影响骨生成稳态。研究表明rankl–rank–opg系统在骨疾病、自身免疫性疾病、器官形成、以及癌症和类风湿性关节炎疾病环境中发挥重要作用。在生理状态下t细胞分泌的rankl在调节性t细胞调控的免疫耐受中发挥着重要作用,研究发现rank可促使胸腺髓质上皮细胞分化和增殖,参与中枢免疫耐受的建立[87]。同时,高水平的rankl与一些自身免疫性疾病中如2型糖尿病(t2dm)及类风湿性关节炎的疾病风险正相关。此外,某些肿瘤细胞可直接产生的rankl或rank,rankl/rank也可以通过激活休眠状态的肿瘤微小病灶,进而直接促进骨肿瘤细胞的生长、存活及转移[88]。rankl/rank系统已成为骨科学、内分泌学、风湿病学、免疫学等众多领域中的最为关键的环节。针对rankl作为治疗靶点已成为药物研究的主要方向。
rankl阻断剂主要有fc-opg、opg-fc、rank-fc、抗人rankl抗体,现均已很好的应用于前期临床及iii期临床研究。其中opg可以结合trail起到活化肿瘤的作用,现已被舍弃。鉴于鼠源性单克隆抗体的多次给药易出现人抗鼠抗体的情况,人源单克隆抗体是进行临床治疗的主要发展趋势。目前,抗人rankl抗体(denosumlab)已应用于iii期临床,它被美国fda批准主要用于治疗和预防绝经后骨质疏松症及乳腺癌和前列腺癌等实体瘤的骨转移导致的骨相关疾病。通过阻断rankl,denosumlab抑制破骨细胞的功能以及同时阻止骨形成与骨吸收,有效的降低了骨折发生的风险。
本次研究中采用了rank的胞外端部分(rank-n)结合rankl而不向下游传递信号这样一个新的策略阻断rankl-rank信号通路。本次研究构建了rank-n巴斯德毕赤酵母表达系统,选择巴斯德毕赤酵母这样一个表达系统是因为它既具有原核系统易于操作,可以以较低成本获得大量蛋白,又具有真核表达系统的翻译后修饰更有可能获得功能活性的蛋白。研究中首先对密码子进行巴斯德毕赤酵母优化,随后进行rank-ppic9k-gs115工程菌的筛选,获得了在6mg/lg418的板子上生长的菌株。有些研究报道发现增加筛选抗生素浓度及对密码子进行巴斯德毕赤酵母优化可有效的增加外源蛋白的表达量[8]。但在我们实验中当遗传霉素g418浓度增加到4mg/l、6mg/l时,两者的外源蛋白表达水平并没有明显差异(图2e)。这可能与蛋白质的稳定性和折叠效率低有关[83],也可能是因为g418筛选浓度和蛋白表达不一定完全线性相关。
rank-ppic9k-gs115工程菌(mus+strain,甲醇利用慢型)在实验室摇瓶中采用1/5摇瓶体积的培养体系,每12小时1%甲醇添加量的诱导表达条件实现了高效与稳定的表达,蛋白表达量>30mg/l,为下一步大规模表达纯化工作奠定了实验基础。
在建立5l大规模发酵中试研究中,并没有获得高表达目的蛋白的菌液上清。通过mm与md平板鉴定发现表达菌株为甲醇利用慢型酵母菌(图3a)。在发酵实验过程中出现od值增加不明显,目的蛋白表达不高的问题。尝试了改变ph、降低诱导温度、延长诱导时间、增加溶氧量、调整初始诱导时的甲醇速率等方法均收效甚微。查阅文献发现虽然甲醇利用慢型菌株可以获得更高的外源蛋白表达量,但过渡转换过程中饥饿培养造成的菌体死亡也会更加影响菌株靶蛋白的表达[84,85]。另外,鉴于在摇瓶中目的蛋白高表达而在高密度发酵中低表达的现象,首先应考虑小分子蛋白的稳定性不高易于降解的问题。特别是在高密度发酵中菌体密度较大,蛋白酶分泌会增加,极易造成对目的蛋白的降解。而出现摇瓶中目的蛋白高表达的原因可能为摇瓶表达时菌密度不高,进而分泌的蛋白酶累积量也不高;其次,摇瓶培养基中含有氮源可部分消除蛋白酶对自身蛋白的氮降解。相应的优化策略有:首先考虑在诱导过程中加入丝氨酸和天冬氨酸型蛋白酶抑制剂pmsf、edta、使用富含氨基酸和抗氧化剂的培养基、与rshpi-1a蛋白酶抑制剂的共表达抑制蛋白酶降解的方法;或者使用smd1163、smd1165、smd1168蛋白酶缺陷菌株、对目的蛋白本身的蛋白酶降解位点进行定点突变[38-40]。这些都是在将来发酵研究中需要优化的地方。
与许多tnfrsf成员类似,rank是一个糖基化蛋白,rank胞外段有3个n-糖基化位点(asn、ser、thr)[3],而酵母中糖基化修饰与人不同,rank胞外段糖基化位点是否影响其在酵母中表达和功能还需进一步研究。
超滤-sephadexg-50凝胶过滤层析-qsepharoseff阴离子交换层析三步法纯化是一个简单可线性化放大高效的纯化方案。在后续的实验中还需要进一步提高蛋白纯度,如改换新的纯化缓冲体系、利用阴阳离子交换纯化等;同时应增强目的蛋白与阴离子交换柱的结合力以减少纯化中的蛋白损失提高得率。
我们在细胞水平证明了我们纯化获得的rank-n是具有阻断人rankl结合rank的活性,并且我们利用biacore-t100系统对rank-n与人rankl进行了蛋白质相互作用分析,测定两者的亲和解离常数kd为2.624e-6m,进一步证明了rank-n是可以高亲和力结合人rankl的。我们利用pdx结直肠癌小鼠移植瘤模型验证了rank-n能够抑制结直肠癌的生长,进一步说明我们纯化的rank-n蛋白在体内也是有功能的。这些实验提示阻断rank信号不仅可以治疗骨相关疾病,还可能直接抑制结直肠癌肿瘤细胞的生长,这是之前没有报道过的。实验表明我们的rank-n蛋白具有很广的临床应用前景。目前实验还需要进一步的完善:1.rank-n治疗体内肿瘤生长的具体机制还需要进一步研究。2.建立大规模发酵中试研究的方法,摸索最适发酵条件实现工业化生产。3.利用pdxdlbcl小鼠移植瘤模型及急性肠炎模型验证体内功能并进行机制研究。
在本发明中,采用巴斯德毕赤酵母表达系统成功表达出人源rank-n胞外段蛋白。本发明建立简单而有效的纯化方案,获得纯度>95%的人源rank胞外段蛋白,并对其生物活性进行一系列体外体内实验评估,发现了rank-n可以在体外抑制破骨细胞的分化,从而抑制骨质疏松;体外抑制弥漫型大b细胞淋巴瘤的存活和生长;体内抑制结直肠癌的生长。人源rank-n胞外段蛋白经体内外实验证明,具有直接抑制破骨细胞分化,抗肿瘤细胞生长的作用,可能用于骨质疏松治疗,各种肿瘤生物治疗,具有潜在的临床应用价值。
本领域技术人员应理解,以上实施例仅是示例性实施例,在不背离本发明的精神和范围的情况下,可以进行多种变化、替换以及改变。
参考文献:
1[1]wongbr,josienr,leesy,etal.thetraffamilyofsignaltransducersmediatesnf-kappabactivationbythetrancereceptor[j].jbiolchem,1998,273(43):28355-28359
2[2]hofbauerlc,khoslas,dunstancr,etal.therolesofosteoprotegerinandosteoprotegerinligandintheparacrineregulationofboneresorption[j].jboneminerres,2000,15(1):2-12.
3[3]andersondm,maraskovskye,billingsleywl,etal.ahomologueofthetnfreceptoranditsligandenhancet-cellgrowthanddendritic-cellfunction[j].nature,1997,390(6656):175-179
4[4]minjk,kimym,kimym,etal.vascularendothelialgrowthfactorup-regulatesexpressionofreceptoractivatorofnf-kappab(rank)inendothelialcells.concomitantincreaseofangiogenicresponsestorankligand[j].jbiolchem,2003.278(41):39548-39557.
5[5]lamj,nelsonca,rossfp,etal.crystalstructureofthetrance/ranklcytokinerevealsdeterminantsofreceptor-ligandspecificity[j].jclininvest,2001,108(7):971-979.
6[6]tanost,sflomosg,echeverriapc,etal.progesterone/ranklisamajorregulatoryaxisinthehumanbreast[j].scitranslmed,2013,5(182):182ra55.
7[7]mabbottna,donaldsonds,ohnoh,etal.microfold(m)cells:importantimmunosurveillancepostsintheintestinalepithelium[j].mucosalimmunol,2013,6(4):666-677.
8[8]akiyamat,shinzawam,qinj,etal.regulationsofgeneexpressioninmedullarythymicepithelialcellsrequiredforpreventingtheonsetofautoimmunediseases[j].frontimmunol,2013,4:249.
9[9]zhaor,chennn,zhouxw,etal.exogenousifn-betaregulatestherankl-c-fos-ifn-betasignalingpathwayinthecollagenantibody-inducedarthritismodel[j].jtranslmed,2014,12(1):330.
10[10]takayanagih,ogasawarak,hidas,etal.t-cell-mediatedregulationofosteoclastogenesisbysignallingcross-talkbetweenranklandifn-gamma[j].nature,2000,408(6812):600-605.
11[11]walshmc,kimn,kadonoy,etal.osteoimmunology:interplaybetweentheimmunesystemandbonemetabolism[j].annurevimmunol,2006,24:33-63.
12[12]greenea,choiy,flavellra.pancreaticlymphnode-derivedcd4(+)cd25(+)tregcells:highlypotentregulatorsofdiabetesthatrequiretrance-ranksignals[j].immunity,2002,16(2):183-191.
13[13]totsukat,kanait,nemotoy,etal.rank-ranklsignalingpathwayiscriticallyinvolvedinthefunctionofcd4+cd25+regulatorytcellsinchroniccolitis[j].jimmunol,2009,182(10):6079-6087.
14[14]khanis,mouchessml,zhuml,etal.enhancementofananti-tumorimmuneresponsebytransientblockadeofcentraltcelltolerance[j].jexpmed,2014,211(5):761-768.
15[15]pearsern,sordilloem,yaccobys,etal.multiplemyelomadisruptsthetrance/osteoprotegerincytokineaxistotriggerbonedestructionandpromotetumorprogression[j].procnatlacadsciusa,2001,98(20):11581-11586.
16[16]yaccobys,pearsern,johnsoncl,etal.myelomainteractswiththebonemarrowmicroenvironmenttoinduceosteoclastogenesisandisdependentonosteoclastactivity[j].brjhaematol,2002,116(2):278-290.
17[17]baud'huinm,duplombl,teletcheas,etal.osteoprotegerin:multiplepartnersformultiplefunctions[j].cytokinegrowthfactorrev,2013,24(5):401-409.
18[18]hsuh,laceydl,dunstancr,etal.tumornecrosisfactorreceptorfamilymemberrankmediatesosteoclastdifferentiationandactivationinducedbyosteoprotegerinligand[j].procnatlacadsciusa,1999,96(7):3540-3545.
19[19]bargmanr,poshamr,boskeyal,etal.comparableoutcomesinfracturereductionandbonepropertieswithranklinhibitionandalendronatetreatmentinamousemodelofosteogenesisimperfecta[j].osteoporosint.,2012,23(3):1141-1150.
20[20]sordilloem,pearsern.rank-fc:atherapeuticantagonistforrank-linmyeloma[j].cancer,2003,97(3suppl):802-812.
21[21]schmiedelbj,scheibleca,nueblingt,etal.ranklexpression,function,andtherapeutictargetinginmultiplemyelomaandchroniclymphocyticleukemia[j].cancerres,2013,73(2):683-694.
22[22]akiyamat,choongpf,dasscr.rank-fcinhibitsmalignancyviainhibitingerkactivationandevokingcaspase-3-mediatedanoikisinhumanosteosarcomacells[j].clinexpmetastasis,2010,27(4):207-215.
23[23]feeleybt,liunq,conduahah,etal.mixedmetastaticlungcancerlesionsinboneareinhibitedbynogginoverexpressionandrank:fcadministration[j].jboneminerres,2006,21(10):1571-1580.
24[24]hollandpm,millerr,jonesj,etal.combinedtherapywiththeranklinhibitorrank-fcandrhapo2l/trail/dulanerminreducesbonelesionsandskeletaltumorburdeninamodelofbreastcancerskeletalmetastasis[j].cancerbiolther,2010,9(7):539-550.
25[25]zhangj,daij,yaoz,etal.solublereceptoractivatorofnuclearfactorkappabfcdiminishesprostatecancerprogressioninbone[j].cancerres,2003,63(22):7883-7890.
26[26]silvai,brancojc.rank/rankl/opg:literaturereview[j].actareumatolport,2011,36(3):209-218.
27[27]voglt,gliedera.regulationofpichiapastorispromotersanditsconsequencesforproteinproduction[j].nbiotechnol,2013,30(4):385-404.
28[28]wertenmw,vandenboschtj,windrd,etal.high-yieldsecretionofrecombinantgelatinsbypichiapastoris[j].yeast,1999,15(11):1087-1096.
29[29]kuprijanova,schaepes,aehlem,etal.improvingcultivationprocessesforrecombinantproteinproduction[j].bioprocessbiosysteng,2012,35(3):333-340.
30[30]creggjm,vedvickts,raschkewc.recentadvancesintheexpressionofforeigngenesinpichiapastoris[j].biotechnology(ny),1993,11(8):905-910.
31[31]creggjm,tolstorukovi,kusaria,etal.expressionintheyeastpichiapastoris.methodsenzymol,2009,463:169-189.
32[32]mattanovichd,callewaertn,rouzép,etal.openaccesstosequence:browsingthepichiapastorisgenome[j].microbcellfact,2009,8:53.
33[33]dalyr,hearnmt.expressionofheterologousproteinsinpichiapastoris:ausefulexperimentaltoolinproteinengineeringandproduction[j].jmolrecognit,2005,18(2):119-138.
34[34]shamloupa.biotechnologyandappliedbiochemistry.editorial[j].biotechnolapplbiochem,2011,58(1):1.
35[35]kjeldsent,petterssonaf,hachm.secretoryexpressionandcharacterizationofinsulininpichiapastoris[j].biotechnolapplbiochem,1999,29(pt1):79-86.
36[36]ghosalkara,sahaiv,srivastavaa.secretoryexpressionofinterferon-alpha2binrecombinantpichiapastorisusingthreedifferentsecretionsignals[j].proteinexprpurif,2008,60(2):103-109.
37[37]lip,anumanthana,gaoxg,etal.expressionofrecombinantproteinsinpichiapastoris[j].applbiochembiotechnol,2007,142(2):105-124
38[38]sinhaj,plantzba,inanm,etal.causesofproteolyticdegradationofsecretedrecombinantproteinsproducedinmethylotrophicyeastpichiapastoris:casestudywithrecombinantovineinterferon-tau[j].biotechnolbioeng,2005,89(1):102-112.
39[39]pyatip,fitchese,gatehouseja.optimisingexpressionoftherecombinantfusionproteinbiopesticideomega-hexatoxin-hv1a/gnainpichiapastoris:sequencemodificationsandasimplemethodforthegenerationofmulti-copystrains[j].jindmicrobiolbiotechnol,2014,41(8):1237-1247.
40[40]fernándeze,toledojr,mansurm,etal.secretionandassemblyofcalicivirus-likeparticlesinhigh-cell-densityyeastfermentations:strategiesbasedonarecombinantnon-specificbpti-kunitz-typeproteaseinhibitor[j].applmicrobiolbiotechnol,2014.
41[41]bretthauerrk,castellinofj.glycosylationofpichiapastoris-derivedproteins[j].biotechnolapplbiochem,1999,30(pt3):193-200.
42[42]hanm,wangw,wangx,etal.enhancedexpressionofrecombinantelastaseinpichiapastoristhroughthesubstitutionofthrforserinasn-xaa-sersequons[j].applbiochembiotechnol,2015,175(1):428-435.
43[43]gemmilltr,trimblerb.overviewofn-ando-linkedoligosaccharidestructuresfoundinvariousyeastspecies[j].biochimbiophysacta,1999,1426(2):227-237.
44[44]kainze,gallmetzera,hatzlc,etal.n-glycanmodificationinaspergillusspecies[j].applenvironmicrobiol,2008,74(4):1076-1086.
45[45]wongbr,josienr,leesy,etal.trance(tumornecrosisfactor[tnf]-relatedactivation-inducedcytokine),anewtnffamilymemberpredominantlyexpressedintcells,isadendriticcell-specificsurvivalfactor[j].jexpmed,1997,186(12):2075-2080.
46[46]ikedat,kasaim,suzukij,etal.multimerizationofthereceptoractivatorofnuclearfactor-kappabligand(rankl)isoformsandregulationofosteoclastogenesis[j].jbiolchem,2003,278(47):47217-47222.
47[47]yasudah,shiman,nakagawan,etal.osteoclastdifferentiationfactorisaligandforosteoprotegerin/osteoclastogenesis-inhibitoryfactorandisidenticaltotrance/rankl[j].procnatlacadsciusa,1998,95(7):3597-3602.
48[48]takahashin,maedak,ishiharaa,etal.regulatorymechanismofosteoclastogenesisbyranklandwntsignals[j].frontbiosci(landmarked),2011,16:21-30.
49[49]nagasawat,kobayashih,kijim,etal.lps-stimulatedhumangingivalfibroblastsinhibitthedifferentiationofmonocytesintoosteoclaststhroughtheproductionofosteoprotegerin[j].clinexpimmunol,2002,130(2):338-344.
50[50]kanazawak,kudoa.self-assembledrankinducesosteoclastogenesisligand-independently[j].jboneminerres,2005,20(11):2053-2060.
51[51]simonetws,laceydl,dunstancr,etal.osteoprotegerin:anovelsecretedproteininvolvedintheregulationofbonedensity[j].cell,1997,89(2):309-319.
52[52]théoleyres,kwantats,vusiop,etal.characterizationofosteoprotegerinbindingtoglycosaminoglycansbysurfaceplasmonresonance:roleintheinteractionswithreceptoractivatorofnuclearfactorkappabligand(rankl)andrank[j].biochembiophysrescommun,2006,347(2):460-467.
53[53]yuntj,chaudharypm,shugl,etal.opg/fdcr-1,atnfreceptorfamilymember,isexpressedinlymphoidcellsandisup-regulatedbyligatingcd40[j].jimmunol,1998,161(11):6113-6121.
54[54]walshmc,choiy.biologyofthetranceaxis[j].cytokinegrowthfactorrev,2003,14(3-4):251-263.
55[55]kimd,mebiusre,macmickingjd,etal.regulationofperipherallymphnodegenesisbythetumornecrosisfactorfamilymembertrance[j].jexpmed,2000,192(10):1467-1478.
56[56]knoopka,butlerbr,kumarn,etal.distinctdevelopmentalrequirementsforisolatedlymphoidfollicleformationinthesmallandlargeintestine:ranklisessentialonlyinthesmallintestine[j].amjpathol,2011,179(4):1861-1871.
57[57]caoy,bonizzig,seagrovestn,etal.ikkalphaprovidesanessentiallinkbetweenranksignalingandcyclind1expressionduringmammaryglanddevelopment[j].cell,2001,107(6):763-775.
58[58]akiyamat,shimoy,yanaih,etal.thetumornecrosisfactorfamilyreceptorsrankandcd40cooperativelyestablishthethymicmedullarymicroenvironmentandself-tolerance[j].immunity,2008,29(3):423-437.
59[59]cowanje,parnellsm,nakamurak,etal.thethymicmedullaisrequiredforfoxp3+regulatorybutnotconventionalcd4+thymocytedevelopment[j].jexpmed,2013,210(4):675-681.
60[60]robertsna,whiteaj,jenkinsonwe,etal.ranksignalinglinksthedevelopmentofinvariantgammadeltatcellprogenitorsandaire(+)medullaryepithelium[j].immunity,2012mar,36(3):427-437.
61[61]jenkinsonsr,williamsja,jeonh,etal.traf3enforcestherequirementfortcellcross-talkinthymicmedullaryepithelialdevelopment[j].procnatlacadsciusa,2013,110(52):21107-21112.
62[62]boycebf.advancesintheregulationofosteoclastsandosteoclastfunctions[j].jdentres,2013,92(10):860-867.
63[63]boylewj,simonetws,laceydl.osteoclastdifferentiationandactivation[j].nature,2003,423(6937):337-342.
64[64]ishidan,hayashik,hoshijimam,etal.largescalegeneexpressionanalysisofosteoclastogenesisinvitroandelucidationofnfat2asakeyregulator[j].jbiolchem,2002,277(43):41147-41156.
65[65]franzosog,carlsonl,xingl,etal.requirementfornf-kappabinosteoclastandb-celldevelopment[j].genesdev,1997,11(24):3482-3496.
66[66]kongyy,yoshidah,sarosii,etal.opglisakeyregulatorofosteoclastogenesis,lymphocytedevelopmentandlymph-nodeorganogenesis[j].nature,1999,397(6717):315-323.
67[67]millerre,roudierm,jonesj,etal.rankligandinhibitionplusdocetaxelimprovessurvivalandreducestumorburdeninamurinemodelofprostatecancerbonemetastasis[j].molcancerther,2008,7(7):2160-2169.
68[68]canonjr,roudierm,bryantr,etal.inhibitionofranklblocksskeletaltumorprogressionandimprovessurvivalinamousemodelofbreastcancerbonemetastasis[j].clinexpmetastasis,2008,25(2):119-129.
69[69]vanderkerkenk,deleenheere,shipmanc,etal.recombinantosteoprotegerindecreasestumorburdenandincreasessurvivalinamurinemodelofmultiplemyeloma[j].cancerres,2003,63(2):287-289.
70[70]santinid,perroneg,roatoi,etal.expressionpatternofreceptoractivatorofnfkappab(rank)inaseriesofprimarysolidtumorsandrelatedbonemetastases[j].jcellphysiol,2011,26(3):780-784.
71[71]cheng,sircark,aprikiana,etal.expressionofrankl/rank/opginprimaryandmetastatichumanprostatecancerasmarkersofdiseasestageandfunctionalregulation[j].cancer,2006,107(2):289-298.
72[72]luojl,tanw,riconojm,etal.nuclearcytokine-activatedikkalphacontrolsprostatecancermetastasisbyrepressingmaspin[j].nature,2007,446(7136):690-694.
73[73]fataje1,kongyy,lij,etal.theosteoclastdifferentiationfactorosteoprotegerin-ligandisessentialformammaryglanddevelopment[j].cell,2000,103(1):41-50.
74[74]gonzalez-suareze,jacobap,jonesj,etal.rankligandmediatesprogestin-inducedmammaryepithelialproliferationandcarcinogenesis[j].nature,2010,468(7320):103-107.
75[75]burgesstl,qiany,kaufmans,etal.theligandforosteoprotegerin(opgl)directlyactivatesmatureosteoclasts[j].jcellbiol,1999,145(3):527-538.
76[76]holeni,crossss,neville-webbehl,etal.osteoprotegerin(opg)expressionbybreastcancercellsinvitroandbreasttumoursinvivo--aroleintumourcellsurvival[j]?breastcancerrestreat,2005,92(3):207-215.
77[77]itor,nakayamah,yoshidak,etal.expressionofosteoprotegerincorrelateswithaggressivenessandpoorprognosisofgastriccarcinoma[j].virchowsarch,2003,443(2):146-151.
78[78]bekkerpj,hollowaydl,rasmussenas,etal.asingle-doseplacebo-controlledstudyofamg162,afullyhumanmonoclonalantibodytorankl,inpostmenopausalwomen[j].jboneminerres,2005,20(12):2275-2282.
79[79]lewieckiem.denosumab--anemergingtreatmentforpostmenopausalosteoporosis[j].expertopinbiolther,2010,10(3):467-476.
80[80]smithmr,saadf,colemanr,etal.denosumabandbone-metastasis-freesurvivalinmenwithcastration-resistantprostatecancer:resultsofaphase3,randomised,placebo-controlledtrial[j].lancet,2012,379(9810):39-46.
81[81]chawlas,henshawr,seegerl,etal.safetyandefficacyofdenosumabforadultsandskeletallymatureadolescentswithgiantcelltumourofbone:interimanalysisofanopen-label,parallel-group,phase2study[j].lancetoncol,2013,14(9):901-908.
82[82]chengml,fongl.effectsofrankl-targetedtherapyinimmunityandcancer[j].frontoncol,2014,3:329.
83[83]whytesideg,alcocermj,kumitajr,etal.native-statestabilitydeterminestheextentofdegradationrelativetosecretionofproteinvariantsfrompichiapastoris[j].plosone,2011,6(7):e22692.
84[84]celike,calikp,oliversg.fed-batchmethanolfeedingstrategyforrecombinantproteinproductionbypichiapastorisinthepresenceofco-substratesorbitol[j].yeast,2009,26(9):473-484.
85[85]inanm,meaghermm.non-repressingcarbonsourcesforalcoholoxidase(aox1)promoterofpichiapastoris[j].jbioscibioeng,2001,92(6):585-589.
核苷酸序列表
<110>上海埃文生物科技有限公司
<120>巴斯德毕赤酵母中人rank胞外段蛋白的表达纯化及其应用
<130>2015
<160>6
<170>patentinversion3.3
<210>1
<211>15
<212>dna
<213>人
<400>1
cgcctcgagaaaaga15
<210>2
<211>552
<212>dna
<213>人
<400>2
cagatcgctcctccatgtaccagtgagaagcattatgagcatctgggacggtgctgtaac60
aaatgtgaaccaggaaagtacatgtcttctaaatgcactactacctctgacagtgtatgt120
ctgccctgtggcccggatgaatacttggatagctggaatgaagaagataaatgcttgctg180
cataaagtttgtgatacaggcaaggccctggtggccgtggtcgccggcaacagcacgacc240
ccccggcgctgcgcgtgcacggctgggtaccactggagccaggactgcgagtgctgccgc300
cgcaacaccgagtgcgcgccgggcctgggcgcccagcacccgttgcagctcaacaaggac360
acagtgtgcaaaccttgccttgcaggctacttctctgatgccttttcctccacggacaaa420
tgcagaccctggaccaactgtaccttccttggaaagagagtagaacatcatgggacagag480
aaatccgatgcggtttgcagttcttctctgccagctagaaaaccaccaaatgaaccccat540
gtttacttgccc552
<210>3
<211>19
<212>dna
<213>人
<400>3
taagcggccgcattattaa19
<210>4
<211>15
<212>dna
<213>人工序列
<400>4
cgcctcgagaaaaga15
<210>5
<211>552
<212>dna
<213>人工序列
<400>5
cagattgctcctccatgtaccagtgagaagcattatgagcatctgggaagatgctgtaac60
aaatgtgaaccaggaaagtacatgtcttctaaatgcactactacctctgacagtgtatgt120
ctgccctgtggccctgatgaatacttggattcatggaatgaagaagataaatgcttgcta180
cataaagtttgtgatacaggcaaggccttggtggccgtggtcgccggtaactcaacgacc240
cccaggcgttgcgcttgcacggctgggtaccactggtcacaggactgcgagtgctgtcgt300
cgaaacactgagtgtgctcctggtttaggtgcacaacaccctttgcaactaaacaaggac360
acagtttgtaaaccttgtcttgcaggttatttctctgatgcattttcctccactgacaaa420
tgtagaccctggactaattgtacttttcttggaaagagagtcgaacatcacggtacagag480
aagtccgatgctgtttgtagttcttccttaccagctagaaagccaccaaatgaacctcac540
gtttatttgcca552
<210>6
<211>19
<212>dna
<213>人工序列
<400>6
taagcggccgcattattaa19
- 还没有人留言评论。精彩留言会获得点赞!