穿心莲内酯衍生物及其3,19酯化物在制备抗肝纤维化药物中的应用的制作方法
本发明涉及穿心莲内酯衍生物作为抗肝纤维化药物的应用,具体涉及15-苄亚基-14-脱氧-11,12-脱氢穿心莲内酯衍生物,属医药技术领域。
背景技术:
全球范围内,肝纤维化(hepaticfibrosis,hf)的发病率约为100/10万人,多见于35-50岁的成年人,其中男性多于女性,农村高于城市。hf是指肝细胞受到损害后,细胞外基质(extracellularmatrix,ecm)与结缔组织异常增生的现象。hf会导致肝组织结构和功能发生改变,当细胞外基质发生长时间的持续性累积,纤维增生形成网状间隔阻碍了肝细胞与血液之间正常的物质与能量交换,大量肝细胞出现坏死,形成肝硬化(livercirrhosis,lc)。据世界卫生组织的定义,lc指弥漫性肝脏纤维化并伴有异常结节增生,最终引发肝衰竭和肝癌(hepatocellularcarcinoma,hcc)。临床研究认为hf是一种可逆性的病变,而lc和hcc则是不可逆的高死亡率病变。因此,控制肝纤维化的发生、发展,可有效控制肝硬化、乃至肝癌的发生,对于延长慢性肝炎患者寿命,提高生活质量具有重大的意义。
临床上酒精肝、脂肪肝、病毒性肝炎、自身免疫性疾病和代谢紊乱等都可引发肝纤维化。正常肝脏的ecm处于动态平衡状态。慢性肝损伤发生时,ecm的合成速度远远高于降解速度,大量胶原蛋白交联成网形成纤维膈膜,进一步引发炎症反应,导致恶性循环,刺激纤维组织生成。当有病毒侵入、有毒物质进入和发生代谢紊乱时,受损肝细胞周围聚集大量炎症细胞,部分肝细胞由于受到损伤导致膜的通透性增大,释放出的代谢产物、活性氧和细胞因子能够激活枯否细胞,进一步分泌大量细胞因子,多种细胞因子共同作用并激活hsc转化成为肌成纤维细胞(myofibroblasts,mfs),在快速增殖的同时分泌炎症介质、粘附分子和大量的胶原物质,同时会抑制基质金属蛋白酶(matrixmetalloproteinase,mmp)的表达,促进金属蛋白酶抑制剂(tissueinhibitorofmetalloproteinase,timp)的分泌,迫使大量的emc堆积在肝脏中。mfs主要来自活化的hsc,当发生炎症反应或受到机械损伤时,hsc受旁分泌影响迅速发生活化,细胞体积变大,分裂能力和收缩能力增强,释放大量的促炎因子、促纤维化因子和细胞分裂因子,并上调α-sma的表达,使ecm的分泌量增加。
目前,临床上尚无针对肝纤维化的特效药物,大多仅起到辅助治疗作用,长期使用甚至会加重肝脏负担。研究显示,干扰素通过阻止肝细胞变性和程序性死亡来控制肝纤维化,抑制胶原形成,促进胶原降解来阻碍肝纤维化的进展。核苷类似物如拉米夫定、阿德福韦酯等可以通过抑制病毒dna的复制来降低血清中病毒的载量,从而起到控制致病因素、降低炎症反应和恢复肝脏功能的疗效。但干扰素的治愈率不高,治疗期间副作用较多,长期使用耐药率高且在纤维化后期治疗效果不显著。核苷类似物并不能对肝纤维化有直接的治疗作用。免疫异常是肝病中普遍存在的问题,常使用环孢菌素a、泼尼松、硫唑嘌呤等强免疫抑制剂来缓解过度的炎症反应,但是在用药过程中免疫抑制剂会对肝脏带来沉重的负担,故肝功能不全的患者禁忌使用。绝大多数肝硬化患者的肝功能已经受到严重的损害,加之单独使用免疫抑制剂后治疗效果一般,所以并未广泛使用。临床上常使用抗炎、抗氧化的细胞保护类药物,通过减轻肝脏组织炎症反应,清除肝脏内的活性氧自由基,对抗脂质过氧化、维持肝细胞膜的流动性来保护肝细胞,使其避免受到损伤,如水飞蓟素、马洛替酯等。
然而,由于hf发生发展机制涉及多种细胞因子及细胞内信号分子网络,所以抗肝纤维化药物需针对多靶点、多环节、多途径发挥作用,所以基于中草药有效成分开发抗肝纤维化药物具有显著优势。穿心莲内酯是穿心莲andrographispaniculata(burm.f.)nees中提取的二萜内酯类化合物,是中药穿心莲的主要有效成份之一。临床上主要用于治疗上呼吸道感染、细菌性痢疾等。近年来,穿心莲内酯在抗肿瘤、保肝利胆、抗病毒等方面的应用研究不断深入。穿心莲内酯对多种动物实验性肝损伤具有良好的保护作用。姚青等发现穿心莲内酯对可卡因引起的急性肝脏损害有一定的保护作用,其保肝机制可能与抑制脂质过氧化反应,降低组织中氧自由基的生成有关。visenpk[jethnopharmacol,1993,40(2),131-136]与handas[indianjmedres,1990,92:284-292]皆证明穿心莲内酯对扑热息痛诱导的肝损伤具有保护作用,其中handas的研究还显示穿心莲内酯对半乳糖胺引起的肝中毒具有保护作用。kapila[biochempharmacol,1993,46(1):182-185]等证明了穿心莲内酯、穿心莲内酯甙和新穿心莲内酯对四氯化碳和叔丁基过氧化氢引起的肝中毒具有保护作用。singhap[jethnopharmacol,2007,111(1):13-21]等的研究显示穿心莲内酯对乙醇引起的小鼠肝肾损伤具有一定的保护作用。roydn[toxicolapplpharmacol,2011;250(1):54-68]等的研究证明穿心莲内酯与d-青霉胺联合治疗铜中毒比单一d-青霉胺在抗纤维化及细胞坏死方面效果更佳。宁光等在其申请的专利(cn201010266185.2)中公开了穿心莲内酯作为制备治疗急性肝损伤药物的应用,穿心莲内酯可以显著抑制刀豆素a诱发的肝损伤,抑制刀豆素a引起的肝细胞的凋亡,抑制肝脏的炎症反应。因此,可以用来治疗刀豆素a诱发的肝损伤。
本发明人在前期研究中(cn1978437;cn100999520;cn100999535;cn101003527)获得了大量结构新颖的化合物。本发明人已对该类化合物在抗肿瘤、抗炎、抗hbv、hcv及急性肝损伤保护作用等应用申请了专利保护,进一步通过对15-苄亚基-14-脱氧-11,12-脱氢穿心莲内酯衍生物及其3,19酯化物在抗肝纤维化方面进行活性试验研究。目前未见相关文献报道。
技术实现要素:
本发明人在前期研究成果的基础上,通过对合成的化合物进行抗肝纤维化活性筛选,发现通式1结构的穿心莲内酯衍生物具有显著的预防和治疗肝纤维化作用,高效低毒,具有开发为抗肝纤维化药物的潜力。本发明目的在于提供15-苄亚基-14-脱氧-11,12-脱氢穿心莲内酯衍生物及其3,19酯化物在制备抗肝纤维化药物中的应用。
所述穿心莲内酯衍生物及其3,19酯化物具有通式1所示结构。
其中:r1、r2分别为氢、甲基或者苯基、吡啶基、呋喃基、噻吩基、吡咯基等芳香环、杂芳香环和它们的单取代、多取代物中的一种;r1、r2分别为环己基、环戊基等环状取代基团中的一种;r1、r2可以是相同的取代基团或者不同的取代基团。r3、r4分别为氢、3-吡啶基或ch2ch2cooh、ch2ch2ch2ch2cooh、ch2chchch2cooh、ch2ch2ch2ch2ch2ch2ch2cooh等中的一种或cor5,r5为取代或者未取代的苯基,吡啶基、吡咯基、呋喃基等芳香杂环或环己基、环戊基、环丙基、吗啉基、哌啶基等碳环或者杂环结构以及饱和或者不饱和的c1-c18长度的碳链等。r3、r4可以是相同的取代基团或者不同的取代基团。
优选15-苄亚基-14-脱氧-11,12-脱氢穿心莲内酯衍生物及其3,19酯化物,其为如下化合物:r1,r2各自为氢或苯基、2-甲氧基苯基、3-甲氧基苯基、4-甲氧基苯基、2,3,5-三甲氧基苯基、2-羟基苯基、3-羟基苯基、4-羟基苯基、2-氟苯基、2-氯苯基、2-溴苯基、3-氟苯基、3-氯苯基、3-溴苯基、4-氟苯基、4-氯苯基、4-溴苯基、2-氟-3-甲氧基苯基、3-甲氧基-4-氯苯基、2,4-二氟苯基、2,4-二氯苯基、2,4-二溴苯基、2-氟-4-氯苯基、2-溴-4-氯苯基、3-氟-4-氯苯基、3-溴-4-氯苯基、3,4-二氟苯基、3,4-二氯苯基、3,4二溴苯基、2-氯-4-氟苯基、2-溴-4-氟苯基、3-氯-4-氟苯基、3-溴-4-氟苯基、2-氟-4-溴苯基、2-氯-4-溴苯基、3-氟-4-溴苯基、3-氯-4-溴苯基、2,3,4-三氯苯基、2-甲氧基-4-氯苯基、2-羟基-4-氯苯基、2-羟基-4-甲氧基苯基、3-氨基-4-氯苯基、2-氨基-4-氯苯基,2-硝基-4-氟苯基,2-硝基-4-氯苯基,r1,r2相同或不同但不同时为氢;r3、r4各自为氢;或r3、r4分别为3-吡啶基或ch2ch2cooh、ch2ch2ch2ch2cooh、ch2chchch2cooh、ch2ch2ch2ch2ch2ch2ch2cooh中的一种,或者r3、r4各自为cor5;r5为3-吡啶基或ch2ch2cooh、ch2ch2ch2ch2cooh、ch2chchch2cooh、ch2ch2ch2ch2ch2ch2ch2cooh中的一种,r3、r4选相同或不同的取代基团。
优选化合物为:r1,r2各自为氢或苯基、2-甲氧基苯基、3-甲氧基苯基、4-甲氧基苯基、2,3,5-三甲氧基苯基、2-羟基苯基、3-羟基苯基、4-羟基苯基、2-氟苯基、2-氯苯基、2-溴苯基、3-氟苯基、3-氯苯基、3-溴苯基、4-氟苯基、4-氯苯基、4-溴苯基、2-氟-3-甲氧基苯基、3-甲氧基-4-氯苯基、2,4-二氟苯基、2,4-二氯苯基、2,4-二溴苯基、2-氟-4-氯苯基、2-溴-4-氯苯基、3-氟-4-氯苯基、3-溴-4-氯苯基、3,4-二氟苯基、3,4-二氯苯基、3,4二溴苯基、2-氯-4-氟苯基、2-溴-4-氟苯基、3-氯-4-氟苯基、3-溴-4-氟苯基、2-氟-4-溴苯基、2-氯-4-溴苯基、3-氟-4-溴苯基、3-氯-4-溴苯基、2,3,4-三氯苯基、2-甲氧基-4-氯苯基、2-羟基-4-氯苯基、2-羟基-4-甲氧基苯基、3-氨基-4-氯苯基、2-氨基-4-氯苯基,2-硝基-4-氟苯基,2-硝基-4-氯苯基,但r1,r2不同;r3、r4各自为氢;或r3、r4分别为ch2ch2cooh、ch2ch2ch2ch2cooh、ch2chchch2cooh、ch2ch2ch2ch2ch2ch2ch2cooh中的一种,或者r3、r4各自为cor5,r5为3-吡啶基或ch2ch2cooh,r3、r4选相同取代基团。
更优选化合物为:当r1,r2其中之一为氢时,r1,r2其中之一选如下基团:苯基、2-甲氧基苯基、3-甲氧基苯基、4-甲氧基苯基、2-氟苯基、2-氯苯基、2-溴苯基、3-氟苯基、3-氯苯基、3-溴苯基、4-氟苯基、4-氯苯基、4-溴苯基、2-氟-3-甲氧基苯基、3-甲氧基-4-氯苯基、2,4-二氟苯基、2,4-二氯苯基、2,4-二溴苯基、2-氟-4-氯苯基、2-溴-4-氯苯基、3-氟-4-氯苯基、3-溴-4-氯苯基、3,4-二氟苯基、3,4-二氯苯基、3,4二溴苯基、2-氯-4-氟苯基、2-溴-4-氟苯基、3-氯-4-氟苯基、3-溴-4-氟苯基、2-氟-4-溴苯基、2-氯-4-溴苯基、3-氟-4-溴苯基、3-氯-4-溴苯基、2-甲氧基-4-氯苯基;r3、r4各自为氢;或r3、r4分别为ch2ch2cooh、ch2ch2ch2ch2cooh、ch2chchch2cooh、ch2ch2ch2ch2ch2ch2ch2cooh中的一种,或者r3、r4各自为cor5,r5为3-吡啶基或ch2ch2cooh,r3、r4选相同取代基团。
更优选化合物为:
a:r1=h,r2=c6h5,r3=r4=h;
b:r1=h,r2=2-f-c6h4,r3=r4=h;
c:r1=h,r2=2-cl-c6h4,r3=r4=h;
d:r1=h,r2=2-br-c6h4,r3=r4=h;
e:r1=h,r2=3-f-c6h4,r3=r4=h;
f:r1=h,r2=3-cl-c6h4,r3=r4=h;
g:r1=h,r2=3-br-c6h4,r3=r4=h;
h:r1=h,r2=4-cl-c6h4,r3=r4=h;
i:r1=h,r2=4-f-c6h4,r3=r4=h;
j:r1=h,r2=4-br-c6h4,r3=r4=h;
k:r1=h,r2=4-ch3o-c6h4,r3=r4=h;
l:r1=h,r2=2-ch3o-4-cl-c6h3,r3=r4=h;
m:r2=h,r1=2-br-c6h4,r3=r4=h;
n:r2=h,r1=3-cl-c6h4,r3=r4=h;
o:r2=h,r1=2-f-4-cl-c6h3,r3=r4=h;
p:r2=h,r1=2,4-dicl-c6h3,r3=r4=h;
q:r2=h,r1=4-f-c6h4,r3=r4=h;
r:r2=h,r1=c6h5,r3=r4=h;
s:r1=h,r2=3-f-4-cl-c6h3,r3=r4=h;
t:r1=h,r2=2,4-dif-c6h3,r3=r4=h;
u:r1=h,r2=3,4-dicl-c6h3,r3=r4=h;
v:r1=h,r2=4-cl-c6h4,r3=r4=cor5,r5=3-吡啶基;
w:r1=h,r2=4-cl-c6h4,r3=r4=ch2ch2cooh;
x:r1=h,r2=4-cl-c6h4,r3=r4=cor5,r5=ch2ch2cooh;
y:r2=h,r1=4-cl-c6h4,r3=r4=h;
z:r2=h,r1=4-cl-c6h4,r3=r4=cor5,r5=3-吡啶基。
本发明提出的上述化合物其制备方法已在发明专利cn:200510107247.4中公开。为研究所述化合物在制备抗肝纤维化药物中的应用效果,本发明利用人肝星状细胞lx-2,测定本发明化合物对细胞迁移和活化的抑制活性,评价化合物的抗肝纤维化活性;进一步以ccl4诱导的sd大鼠肝纤维化模型、猪血清诱导的wistar大鼠肝纤维化模型和胆管结扎(bdl)致sd大鼠肝纤维化模型为代表,通过测定肝组织胶原含量(masson染色)、hsc活化标志分子α-sma表达水平,评价化合物的抗肝纤维化活性。进一步,通过ccl4模型,系统研究说明了代表性化合物发挥抗肝纤维化作用的主要相关作用环节和机制。
该类化合物的顺、反结构均具有抗肝纤维化活性,以该类化合物为有效药用成份,或该类化合物的各类前药形式,单独或与其它药物组合,按目前各种常规的制药方法和工艺要求,与制药中可以接受的辅助和/或添加成份混合后,制成用于抗肝纤维化的口服型制剂、注射型制剂等各类药物剂型。优选将其制备治疗或预防由毒物、病毒、代谢、免疫、酒精等各类因素导致的肝纤维化药物。
口服型制剂为片剂、丸剂、胶囊、冲剂或糖浆等;注射型制剂包括注射液或冻干粉针剂型等。
本发明优点及创新点:通过活性筛选,确定上述化合物具有明确的抗肝纤维化活性。经实验证明,该类化合物显著抑制肝星状细胞迁移、活化,降低细胞外基质(ecm)相关组分含量,降低肝纤维化大鼠肝组织纤维化水平;显著降低免疫炎症相关因子的水平,并有效抑制免疫炎症反应。抑制肝组织肝星状细胞活化,促进胶原降解,效果显著且优于母体化合物穿心莲内酯(ad),高效低毒。将该类化合物作为活性成份用于制备抗肝纤维化药物,为肝纤维化的治疗和预防提供了新的药物途径,从而扩大了临床用药的可选择范围,具有良好的应用开发前景。填补肝纤维化无理想治疗药物的空白。
附图说明
图1为ad和本发明化合物(15μm)对人肝星状细胞lx-2活力的影响,与ad组比,#p<0.05;
图2为ad和本发明化合物(5μm)抑制人肝星状细胞lx-2迁移的结果(统计结果),与ad组比,#p<0.05;
图3为ad和本发明化合物(5μm)抑制人肝星状细胞lx-2迁移的结果(部分显微图片;×100倍);
图4为本发明部分代表性化合物对ccl4诱导的肝纤维化sd大鼠肝组织纤维化程度的影响(相对胶原面积/%),与模型组比,*p<0.05;与ad组比,#p<0.05;与水飞蓟宾组比,&p<0.05;
图5为本发明部分代表性化合物对ccl4诱导的肝纤维化sd大鼠肝组织纤维化程度的影响(部分masson染色图片;×100倍);
图6为本发明部分代表性化合物对ccl4诱导的肝纤维化sd大鼠肝组织α-平滑肌肌动蛋白(α-sma)表达水平的影响(统计结果),与模型组比,*p<0.05;与ad组比,#p<0.05;与水飞蓟宾组比,&p<0.05;
图7为本发明部分代表性化合物对ccl4诱导的肝纤维化sd大鼠肝组织α-sma表达水平的影响(部分免疫组化图片;×100倍);
图8为本发明部分代表性化合物对猪血清诱导的肝纤维化wistar大鼠肝组织纤维化程度的影响(相对胶原面积/%),与模型组比,*p<0.05;与ad组比,#p<0.05;与水飞蓟宾组比,&p<0.05;
图9为本发明部分代表性化合物对猪血清诱导的肝纤维化wistar大鼠肝组织纤维化程度的影响(部分masson染色图片;×100倍);
图10为本发明部分代表性化合物对猪血清诱导的肝纤维化wistar大鼠肝组织α-sma表达水平的影响(统计结果),与模型组比,*p<0.05;与ad组比,#p<0.05;与水飞蓟宾组比,&p<0.05;
图11为本发明部分代表性化合物对猪血清诱导的肝纤维化wistar大鼠肝组织α-sma表达水平的影响(部分免疫组化图片;×100倍);
图12为本发明部分代表性化合物对总胆管结扎致sd大鼠肝组织纤维化程度的影响(相对胶原面积/%),与模型组比,*p<0.05;与ad组比,#p<0.05;与熊去氧胆酸组比,&p<0.05;
图13为本发明部分代表性化合物对总胆管结扎致sd大鼠肝组织纤维化程度的影响(部分masson染色图片;×100倍);
图14为本发明部分代表性化合物对总胆管结扎致sd大鼠肝组织α-sma表达水平的影响(统计结果),与模型组比,*p<0.05;与ad组比,#p<0.05;与熊去氧胆酸组比,&p<0.05;
图15为本发明部分代表性化合物对总胆管结扎致sd大鼠肝组织α-sma表达水平的影响(部分免疫组化图片;×100倍);
图16为本发明代表化合物h对ccl4诱导的肝纤维化sd大鼠血清中层粘连蛋白(laminin,ln)水平的影响,与模型组比,*p<0.05;与苦参素组比,&p<0.05;
图17为本发明代表化合物h对ccl4诱导的肝纤维化sd大鼠血清中i型胶原(c-ⅰ)水平的影响,与模型组比,*p<0.05;与苦参素组比,&p<0.05;
图18为本发明代表化合物h对ccl4诱导的肝纤维化sd大鼠血清中前iii型胶原(pc-ⅲ)水平的影响,与模型组比,*p<0.05;与ad组比,#p<0.05;与苦参素组比,&p<0.05;
图19为本发明代表化合物h对ccl4诱导的肝纤维化sd大鼠血清中ⅳ型胶原(c-ⅳ)水平的影响,与模型组比,*p<0.05;与苦参素组比,&p<0.05;
图20为本发明代表化合物h对ccl4诱导的肝纤维化sd大鼠血清中白介素-6(il-6)水平的影响,与模型组比,*p<0.05;与ad组比,#p<0.05;与苦参素组比,&p<0.05;
图21为本发明代表化合物h对ccl4诱导的肝纤维化sd大鼠血清中肿瘤坏死因子-α(tnf-α)水平的影响,与模型组比,*p<0.05;与ad组比,#p<0.05;与苦参素组比,&p<0.05;
图22为本发明代表化合物h对ccl4诱导的肝纤维化sd大鼠肝组织中超氧化物歧化酶(sod)水平的影响,与模型组比,*p<0.05;与苦参素组比,&p<0.05;
图23为本发明代表化合物h对ccl4诱导的肝纤维化sd大鼠肝组织中丙二醛(mda)水平的影响,与模型组比,*p<0.05;
图24为本发明代表化合物h对ccl4诱导的肝纤维化sd大鼠肝组织病理切片炎症级别的影响(统计结果),与模型组比,*p<0.05;与ad组比,#p<0.05;与苦参素组比,&p<0.05;
图25为本发明代表化合物h对ccl4诱导的肝纤维化sd大鼠肝组织病理切片炎症级别的影响(h&e染色;×100倍)。
具体实施方案
下面结合具体实施方案来详细阐述本发明。应理解这些实施方案仅用于说明本发明而不用于限制本发明的范围,本发明涉及的化合物不限于实施例中使用的代表性结构,可以更换15位的不同取代基,获得具有抗肝纤维化活性的化合物;还可以用各种导致肝纤维化的原因作为研究对象来得出本发明物具有抗肝纤维化作用。
实施例1本发明化合物抑制人肝星状细胞lx-2迁移作用。
在各种炎症介质、生长因子等细胞因子的刺激下,肝星状细胞迁移到受损肝组织的炎症部位,进而增殖、活化,合成胶原等ecm成分是肝纤维化发生发展的关键。因此,采用划痕损伤法评价本发明化合物抗肝纤维化作用。
1细胞培养和药物处理
采用人肝星状细胞lx-2(由北京北纳创联生物技术研究院提供),与穿心莲内酯比较,研究本发明化合物的体外抗肝纤维化作用。将lx-2细胞培养在含rpmi1640培养液的培养瓶中,培养液中含体积分数10%胎牛血清(浙江天杭生物科技股份有限公司,批号:100524)、100μg/ml链霉素、100iu/ml青霉素,置体积分数5%co2培养箱(德国binder公司)中于饱和湿度、37℃培养。
2mtt法测定细胞毒
将生长对数期的lx-2细胞用0.25%胰蛋白酶消化后,用含体积分数10%胎牛血清的rpmi1640培养基稀释成3.5×105/ml细胞悬液,铺于96孔板(美国costar公司)内,200μl/孔,于37℃,体积分数5%co2培养箱中培养24h,加入含药培养基,药物终浓度为15μm,继续培养48h,加入mtt(5mg/ml),20μl/孔,培养4h,弃上清,加入150μldmso,震荡l0min,用酶标仪测定吸光值。测定波长为570nm,参考波长为450nm。计算化合物作用后的细胞存活率,存活率(%)=药物组a值/细胞对照组a值×100%,结果见附图1。数据利用spss17.0统计学软件进行处理和分析。数据均采用平均值±标准差
3划痕损伤法观察药物对lx-2细胞迁移的影响
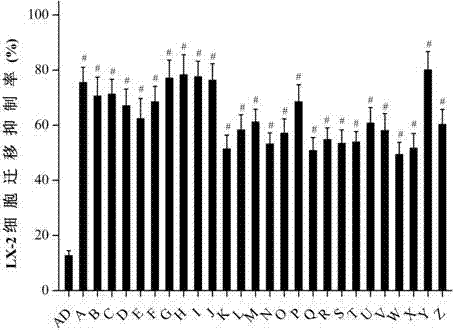
将生长对数期的lx-2细胞用0.25%胰蛋白酶消化后,用含体积分数10%胎牛血清的rpmi1640培养基稀释成1.0×106/ml细胞悬液,铺于96孔板内,每孔200μl。培养12h之后细胞长成融合状态,弃去原培养基,加入血清量为0.5%的培养基再同步化培养12h之后划线,用pbs洗两遍,加入200μl含待测化合物(5μm)的rpmi1640培养基后立即在显微镜下拍照。设3孔重复并且设置对照。培养24h后分别在显微镜下拍照测量。迁移抑制率=1-(给药组0h划痕距离-24h划痕距离)/(空白组0h划痕距离-24h划痕距离)×100%。结果见附图2、3。数据利用spss17.0统计学软件进行处理和分析。数据均采用平均值±标准差
4实验结果
图1结果表明,本发明化合物在15μm浓度下,对人肝星状细胞增殖未表现出明显的抑制作用,且存活率均显著高于母体化合物穿心莲内酯。
结合图1、2、3,结果表明:与ad比,本发明化合物在无毒浓度下,可显著抑制lx-2细胞的迁移,且与ad比,对人肝星状细胞迁移的抑制作用更强,安全指数更高。
实施例2本发明化合物显著降低四氯化碳(ccl4)诱导sd大鼠的肝纤维化程度
ccl4诱导肝纤维化模型是一种传统的经典动物模型,因其在形态学、病理生理学等诸多方面,都与人体肝纤维化有极大的相似性,不仅具有毒物诱导的肝纤维化特征,而且还与乙肝病毒感染后的病理特征相似,能很好地模拟人类肝纤维化的病理变化。受低剂量的ccl4长期刺激后,不但动物表现出的肝功异常与人肝硬化体征十分相似,而且纤维化发生分子机制、损伤后血清标志物、肝组织病理改变与人也非常相似。因此,ccl4诱导肝纤维化模型现被广泛应用于研究肝纤维化发病机制、抗肝纤维化药物筛选、抗肝纤维化药物的作用机制等。
1材料与方法
1)试验动物
清洁级sd大鼠,雄性,体重200±20g,购于湖南斯莱克景达实验动物有限公司。其许可证号是scxk(湘)2011-0003。
2)药品、试剂及其配制
穿心莲内酯由四川什邡市金鑫生物科技有限公司生产(批号:120822),纯度大于99%;本发明化合物由本发明人所在实验室合成,纯度大于99%;药用级低聚羧甲基纤维素钠(cmc-na)由安徽山河药用辅料有限公司生产(批号:131114);苦参素胶囊,产自正大天晴药业公司(批号:国药准字h20010763)。水飞蓟宾胶囊由天津天士力圣特制药有限公司(批号:国药准字h20040299)生产,药物用0.5%cmc-na配成混悬液。ccl4由天津凯基化学试剂公司生产,其它试剂均为市售分析纯。
3)试验方法
sd大鼠适应性喂养3d,按体重随机分组,分出正常组后,其余大鼠进行背部皮下注射含40%ccl4的大豆油造模,首次剂量为4ml/kg,其余剂量为2ml/kg,2次/w。造模4w后,除正常组外,其余大鼠按体重进行分组,8只/组。穿心莲内酯、苦参素、水飞蓟宾和本发明化合物a剂量均为20mg/kg,其他化合物剂量为与a相同的摩尔物质量;模型组和正常组动物给予等量的0.5%cmc-na。第5w、6w造模和给药同时进行,第7w、8w停止造模,只给药。每次灌胃均在早上空腹时进行,每给药10d停药一天。大鼠在末次灌胃前8h更换垫料,严格禁食不禁水。给药后1h腹腔注射3%的戊巴比妥钠(2ml/kg)麻醉剂,采血后迅速完整地剖离肝脏。采集的血液在37℃孵育箱中静置45min后,4℃下3500rpm离心15min,取上层血清,分装备用。取大鼠左叶下半部分肝脏于10倍体积4%多聚甲醛固定液中固定,24h后更新固定液。固定充分后进行病理切片,经masson三色染观察肝脏纤维化程度,image-proplus软件对masson染色切片的拍照结果进行纤维化组织学半定量分析。计算相对胶原面积:(给药组平均面积-正常组平均面积)/(模型组平均面积-正常组平均面积)×100%,结果见附图4、5。利用免疫组织化学法评价肝组织α-sma(hsc活化程度的标志物)表达情况,使用image-proplus对阳性表达进行量化分析,结果见附图6、7。数据利用spss17.0统计学软件进行处理和分析。数据均采用平均值±标准差
2实验结果
结合图4、5、6、7,结果表明:本发明化合物能显著降低动物肝组织的纤维化程度。与ad比,本发明化合物治疗组肝组织的胶原面积显著减少。与阳性参照水飞蓟宾和苦参素比,疗效更突出。同时观察到本发明化合物显著下调肝组织中α-sma的表达水平,与ad比,差异显著,且效果优于阳性药。说明其发挥抗肝纤维化作用与抑制肝星状细胞hsc活化有关。
实施例3本发明化合物显著降低猪血清诱导wistar大鼠的肝纤维化程度
免疫性肝纤维化主要是由肝脏自身出现免疫反应所引起的疾病,而且其他因素(病毒、酒精、血吸虫及某些化学物质等)所致肝纤维化也常伴随不同程度的免疫反应。对实验动物反复腹腔注射异种血清或蛋白(猪血清g、血吸虫血清、人或牛血清白蛋白等)是目前主要的造模方法。该法的独特之处在于异种血清主要通过mhcⅱ分子和炎症因子激活肝干细胞hsc产生免疫应答导致的肝脏损伤,在这个过程中肝纤维化与免疫反应持续存在,能很好地重现其他模型所不能呈现的人类自身免疫性肝损伤所致肝纤维可能的过程和机制,使该模型在抗肝纤维化的免疫性药物的筛选和评估方面具有很大优势。
1材料与方法
1)试验动物
清洁级wistar大鼠,雄性,体重140±20g,购于南疆君科生物工程有限公司,合格证号scxk(辽)2015-0001。
2)药品、试剂及其配制
水飞蓟宾胶囊由天津天士力圣特制药有限公司(批号:国药准字h20040299)生产;猪血清由广州蕊特生物科技有限公司生产(批号:160608)。其他供试药物、化合物同实施例1、2,其它试剂均为市售分析纯;药物配成0.5%的羧甲基纤维素钠(cmc-na)混悬液。
3)试验方法
wistar大鼠适应性喂养3d后,按照体重随机分组,每组6只。除正常组动物外,全部大鼠均进行腹腔注射猪血清,1ml/只,2次/w,持续6w。实验至第8w末结束。灌胃给药,1次/d。每次灌胃均在早上空腹时进行,每给药10d停药一天。预防给药组(h:2.5mg/kg)动物从造模当天开始给药,水飞蓟宾(50mg/kg)、穿心莲内酯(20mg/kg)和本发明化合物h的5mg/kg和10mg/kg剂量组动物均从第5w开始给药,而20mg/kg剂量组动物从第7w开始给药;对照组和模型组灌胃给予0.5%cmc-na。大鼠在末次灌胃前8h更换垫料,严格禁食不禁水。给药后1h腹腔注射3%的戊巴比妥钠(2ml/kg)麻醉剂,采血后迅速完整地剖离肝脏并拍照。采集的血液在37℃孵育箱中静置45min后,4℃下3500rpm离心15min,取上层血清,保存备用。取大鼠左叶下半部分肝脏于10倍体积4%多聚甲醛固定液中固定,24h后更新固定液。固定充分后进行病理切片,经masson三色染观察肝脏纤维化程度,image-proplus软件对masson染色切片的拍照结果进行纤维化组织学半定量分析。计算相对胶原面积:(给药组平均面积-正常组平均面积)/(模型组平均面积-正常组平均面积)×100%,结果见附图8、9。利用免疫组织化学法评价肝组织α-sma(hsc活化程度的标志物)表达情况,使用image-proplus对阳性表达进行量化分析,结果见附图10、11。数据利用spss17.0统计学软件进行处理和分析。数据均采用平均值±标准差
2实验结果
结合图8、9、10、11,结果表明:在猪血清诱导的肝纤维化模型上,本发明化合物表现出良好的抗肝纤维化作用。无论预防给药还是治疗给药均显著减轻了大鼠的肝纤维化程度。效果优于ad和水飞蓟宾(50mg/kg)处理组,即使造模6w后给药,仅给药2w也取得了良好的治疗效果。同时观察到本发明化合物显著下调肝组织中α-sma的表达水平,与ad比,差异显著,且效果优于阳性药。说明其发挥抗肝纤维化作用与抑制肝星状细胞hsc活化有关。
实施例4本发明化合物显著减轻总胆管结扎的sd大鼠肝纤维化程度
1材料与方法
1)试验动物
清洁级sd大鼠,雄性,体重200±20g,购于湖南斯莱克景达实验动物有限公司。其许可证号是scxk(湘)2016-0002。
2)药品、试剂及其配制
熊去氧胆酸由上海信谊药厂有限公司(批号:国药准字h31021875)生产;其他供试药物、化合物同实施例1、2,其它试剂均为市售分析纯;药物配成0.5%的羧甲基纤维素钠(cmc-na)混悬液。
3)试验方法
sd大鼠适应性喂养3d后,按照体重随机分组:假手术对照组、模型组、ad对照组(20mg/kg)、熊去氧胆酸对照组(25mg/kg)和各本发明化合物组,每组4只。假手术组和模型组灌胃给予0.5%cmc-na,其余各给药组给予0.5%cmc-na混悬的相应药物,给药4w结束。术前12h更换垫料,严格禁食不禁水,灌胃前2h肌肉注射青霉素8万u/只,灌胃后1h腹腔注射3%的戊巴比妥钠(2ml/kg),麻醉生效后,仰位固定大鼠四肢,剪毛,碘酒消毒皮肤,铺洞巾,沿腹中线开腹,沿胃向下找到并向上牵引十二指肠,游离胆总管,距近肝门部0.5cm处,以4/0号丝线双重结扎并离断胆总管,检查无出血及胆漏情况后,以3/0号丝线连续缝针法逐层关腹,碘酒消毒伤口,术后保暖至清醒,假手术组只进行麻醉、开腹、游离胆总管,不结扎、不离断胆总管。每次灌胃均在早上空腹时进行,每给药10d停药一天。大鼠在末次灌胃前12h更换垫料,严格禁食不禁水。给药后1h腹腔注射3%的戊巴比妥钠(2ml/kg)麻醉剂,采血后迅速完整地剖离肝脏并拍照。血液离心后,取上清,保存备用。取大鼠左叶下半部分肝脏于10倍体积4%多聚甲醛固定液中固定,24h后更新固定液。固定充分后进行病理切片,经masson三色染观察肝脏纤维化程度,image-proplus软件对masson染色切片的拍照结果进行纤维化组织学半定量分析。计算相对胶原面积:(给药组平均面积-正常组平均面积)/(模型组平均面积-正常组平均面积)×100%,结果见附图12、13。利用免疫组织化学法评价肝组织α-sma(hsc活化程度的标志物)表达情况,使用image-proplus对阳性表达进行量化分析,结果见附图14、15。数据利用spss17.0统计学软件进行处理和分析。数据均采用平均值±标准差
2实验结果
结合图12、13、14、15,结果表明:在胆管阻塞型肝纤维化模型上,本发明化合物抗肝纤维化作用均显著强于ad和阳性药处理。同时观察到本发明化合物显著下调肝组织中α-sma的表达水平,与ad比,差异显著,且效果优于阳性药。说明其发挥抗肝纤维化作用与抑制肝星状细胞hsc活化有关。
实施例5本发明化合物对四氯化碳(ccl4)诱导sd大鼠肝纤维化动物血清胶原指标的影响
正常肝组织中,胶原物质是窦间隙基质膜的重要组成成分,以c-ⅰ和c-ⅳ为主。当肝脏受到损伤后,大量胶原物质和糖蛋白被转录、翻译和组装,以ln、c-ⅰ和c-ⅲ为主。临床上,血清ln、c-ⅰ、pc-ⅲ和c-ⅳ水平是纤维化患者诊断的重要指标。
1材料与方法
同实施例2。
以本发明化合物h为代表,采用elisa方法测定动物血清中ln、c-ⅰ、pc-ⅲ和c-ⅳ水平。血清中胶原指标变化见附图16-19。数据利用spss17.0统计学软件进行处理和分析。数据均采用平均值±标准差
2实验结果
结果显示,与正常组相比,模型组中ln与三种胶原含量均大幅提高;各治疗组显著降低肝组织中ln含量,本发明化合物h高剂量组和阳性药苦参组相比有显著性差异;苦参、ad和本发明化合物均显著降低了c-ⅰ含量,h呈剂量依赖性降低c-ⅰ水平;h低、高剂量显著降低组织内pc-ⅲ水平,给药浓度为20mg/kg时,与苦参组和ad组相比有显著性意义;苦参、ad和化合物h显著降低肝组织中c-ⅳ的含量,h呈剂量依赖性降低血清c-ⅳ水平。
结合图16、17、18、19,结果表明:本发明化合物在保持了母体化合物ad的大幅降低ln、c-ⅰ和c-ⅳ水平的基础上,显著提高了对pc-iii的抑制活性。其中本发明化合物在20mg/kg剂量使ln、pc-ⅲ、c-ⅳ降低到正常水平。并且化合物h在5mg/kg剂量对pc-ⅲ、c-ⅳ的下调程度与ad在20mg/kg剂量时的相当。
实施例6本发明化合物对四氯化碳(ccl4)诱导sd大鼠肝纤维化动物血清il-6和tnf-α水平的影响
活化的hsc可诱导巨噬细胞产生大量的tnf-α,继续参与hsc的分化,同时还会扩大肝脏内炎症反应。il-6是促纤维化因子之一,参与肝组织内炎症反应、脂质过氧化、细胞凋亡与再生等复杂的生理过程,也是nf-κb下游的效应分子。1材料与方法
同实施例2。
以本发明化合物h为代表,采用elisa方法测定动物血清中il-6和tnf-α水平。结果见附图20、21。数据利用spss17.0统计学软件进行处理和分析。数据均采用平均值±标准差
2实验结果
结合图20、21,结果表明:三个剂量的化合物h都使肝纤维化大鼠血清中tnf-α和il-6水平大幅降低,高剂量组效果最为显著,在给药剂量为20mg/kg时血清中il-6和tnf-α的含量与正常组动物水平相当。结果提示:本发明化合物的抗肝纤维化作用与抑制tnf-α、il-6的表达密切相关,通过降低tnf-α和il-6的含量,阻碍hsc分化成为mfc,并通过抑制nf-κb信号通路激活降低了炎症反应。
实施例7本发明化合物对四氯化碳(ccl4)诱导sd大鼠肝纤维化动物肝组织中sod和mda水平的影响
sod和mda是评价脂质过氧化的重要指标。sod是抗氧化剂,不仅可以抑制因自由基启动导致的脂质过氧化,还可以清除自由基以保护生物膜的完整性,是机体内抗氧化能力的敏感指标。脂质过氧化会产生大量的mda,它的含量与组织内过氧化的损伤程度成正比。
1材料与方法
同实施例2。
以本发明化合物h为代表,测定动物肝组织中sod、mda水平。结果见附图22,23。数据利用spss17.0统计学软件进行处理和分析。数据均采用平均值±标准差
2实验结果
结合图22、23,结果表明:本发明化合物5、10、20mg/kg三个剂量组sod水平均大幅升高,并接近正常水平,10、20mg/kg两个剂量组mda水平均大幅降低,并接近正常值。说明本发明化合物保持了ad的强抗脂质过氧化能力。
实施例8本发明化合物大幅改善四氯化碳(ccl4)诱导sd大鼠肝纤维化动物肝组织炎症状态
1材料与方法
同实施例2。
以本发明化合物h为代表,通过h&e染色观察分析本发明化合物对肝组织免疫炎症状态的改善情况,结果见附图24、25。数据利用spss17.0统计学软件进行处理和分析。数据均采用平均值±标准差
2实验结果
结合图24、25,结果表明:正常组大鼠的肝小叶结构完整、肝细胞索排列整齐,没有异变发生且维管区正常;肝细胞未见变形、坏死,无纤维结缔产生。模型组大鼠肝细胞索排列混乱,核质比明显增大并伴随可见点、片状及灶状坏死区,淋巴细胞浸润严重,肝小叶结构严重受损无法辨别。与模型组相比,给药组动物肝损伤均得到不同程度的改善,尤其是本发明化合物h的中、高剂量治疗组最为显著,肝细胞仅有轻微肿大,肝细胞索的结构正常,明显优于阳性药苦参治疗组和ad治疗组。
- 还没有人留言评论。精彩留言会获得点赞!