一种呱西替柳晶型化合物及其制备方法与流程
本发明属于医药
技术领域:
,涉及一种呱西替柳晶型化合物及其制备方法。
背景技术:
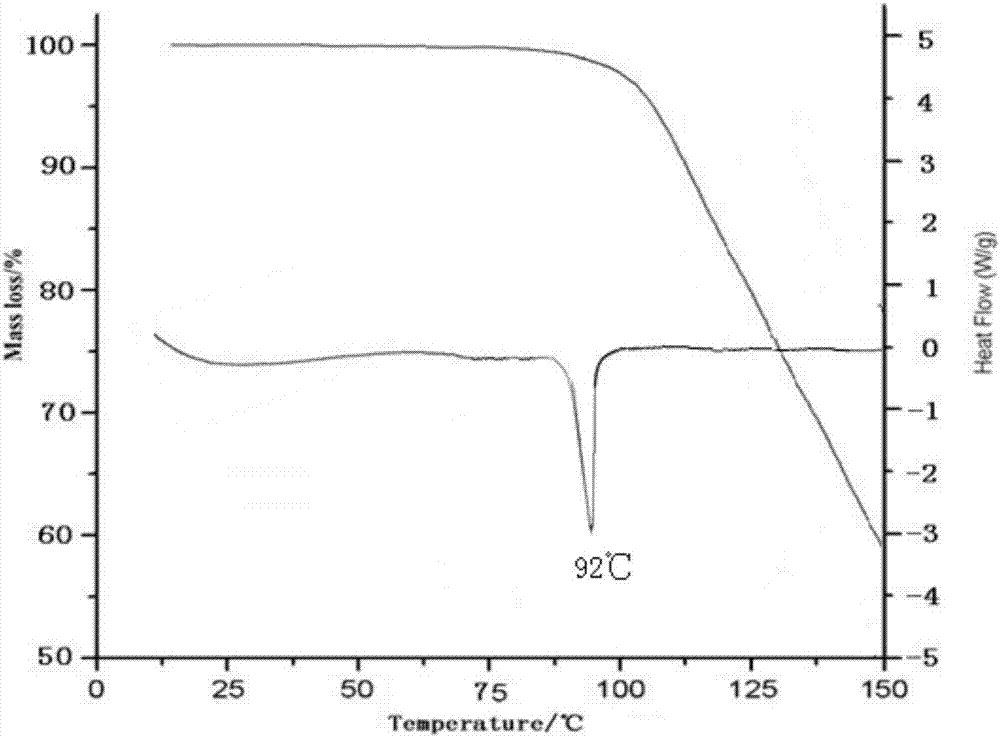
:呱西替柳,是水杨酸类衍生物,结构是如下。呱西替柳1981年在意大利上市,是一种解热镇痛药,能延缓激肽所引起的支气管痉挛,抑制前列腺素PG的合成,稳定溶媒体膜,具有消炎、解热、镇痛的作用。此外,它能使黏液溶解,故而有化痰祛痰作用,及其适用于婴幼儿。呱西替柳在临床上用于治疗伴有发热、疼痛的急慢性呼吸道炎症及粘膜炎症等。呱西替柳又名呱西替沙,醋柳愈酯,醋柳酯,为白色结晶性粉末,几乎无臭,无味。易溶于三氯甲烷、苯、可溶于热乙醇、无水乙醚,不溶于水。一个原料药的不同晶型可以有不同的化学和物理特性,包括熔点、化学反应性、表观溶解度、溶解速率、光学和机械性质、蒸气压和密度。这些特性可以直接影响原料药和制剂的处理和/或生产,并且会影响制剂的稳定性、溶解度和生物利用度。当化合物存在多晶型时,由于特定多晶型物具有特异性的热力学性质和稳定性,因此在制备的过程中,了解在各个剂型中应用的化合物的晶型是重要的,以保证生产过程应用相同形态的药物活性化合物。因此,保持药物活性化合物是单一的晶型或是一些晶型的已知混合物是必要的。现有技术中已有大量的文献及专利报道了呱西替柳的合成方法,大多数采用乙醇重结晶或精制,得到晶体:如《超声波作用下合成药物呱西替柳》医药农药,2009年第02期,陈小全,仇玉芹等;《呱西替柳的催化合成》武汉化工学院学报,2001年第23卷第4期,陈立钦,白崇荣,高锁;《呱西替柳的合成》中国医药工业杂志,1990年21卷第9期,孙长安,黄建军;《呱西替柳的合成工艺改进》华西药学杂志,1998年13卷第3期,张大永,徐鸣夏;《呱西替柳的合成工艺改进》王秋芬,郑更修等;《呱西替柳合成方法的改进》黑龙江大学自然科学学报,1993年第10卷第1期,赵书清,邢有权等;《呱西替柳合成方法的改进》海军医高专学报,1997年第19卷第3期,祁铁流,薛荣,韦玮;CN104744261A,一种呱西替柳的生产方法,张云。也有使用CHCl3析出晶体:如《醋柳愈酯的相转移催化合成》天津化工,2001年第1期,何明华,林家逊。103102271B公开了一种呱西替柳的工业化制备方法,其创新在于向反应液中滴加溶剂甲醇、乙醇、异丙醇、正丙醇或异丁醇,冷却直接析出产品,是将常规处理中把反应液倒入冰水中和再次用乙醇重结晶两步合并一步,简化操作,提高了安全性。本申请进行了大量的试验,发现采用现有技术的方法用乙醇或CHCl3重结晶或采用CN103102271B的方法用甲醇、乙醇、异丙醇、正丙醇或异丁醇冷却析晶,或将甲醇、乙醇、异丙醇、正丙醇、异丁醇及CHCl3两种、三种或多种溶剂混合结晶或降温析晶或采用现有技术的工艺制备晶体(本发明采用的溶剂组合除外),均得到晶型I化合物,且晶型I化合物均存在水溶性不好(<0.013mg/ml),有关物质含量高(>0.4%),单杂含量高(>0.1%)。现有技术呱西替柳水溶解性极低,其制剂的生物利用度好坏直接影响其疗效,且呱西替柳各杂质结构中均含有水杨酸(或乙酰水杨酸)和愈创木酚结构单元,其分解产物均为低毒性物质,直接影响药品质量和用药安全。本发明经过大量的试验研究,采用新的结晶方法,得到了一种新的呱西替柳晶型II化合物,本发明的呱西替柳晶型II化合物提高了水溶性且具有较高的稳定性,有关物质含量显著降低,制备方法简单易操作,制成药物组合物后用药安全性大大提高,非常适合临床应用。技术实现要素:本发明的首要发明目的在于提出了一种呱西替柳晶型化合物及其制备方法。为了实现本发明的目的,采用的技术方案为:本发明提供一种呱西替柳晶型化合物,该化合物以2θ±0.2°衍射角表示的X-射线粉末衍射图谱在9.16°、9.53°、10.84°、11.50°、12.84°、13.79°、14.32°、14.79°、15.47°、16.26°、18.45°、18.92°、19.65°、20.54°、21.53°、21.83°、22.56°、23.12°、23.79°、25.44°、26.55°、27.36°和28.46°处显示有特征衍射峰。本发明提供的呱西替柳晶型化合物使用Cu-Kα射线测量得到的X-射线粉末衍射图如图1所示。本发明还提供了一种呱西替柳晶型化合物的制备方法,具体步骤为:a)将呱西替柳粗品溶于混合溶剂A,溶液搅拌加热后使呱西替柳粗品全部溶解;b)将上述溶液降温至-5℃~0℃,向溶液中按1.0~2.0mL/min的流速加入甲醇至出晶,继续降温至-10℃~-5℃,保温搅拌养晶至析晶完全;c)抽滤,收集晶体,真空干燥,得到呱西替柳结晶。优选地,步骤a)中,所述混合溶剂A为氯仿和甲醇的混合溶剂,氯仿和甲醇的体积比为2~3:1;呱西替柳和混合溶剂A的质量体积比为1:10~20。优选地,步骤b)中,所述的甲醇与混合溶剂A的体积比为2~5:1。更优选地,步骤b)中,降温幅度为每10分钟1℃~5℃,养晶温度为-10℃~-5℃,养晶时间为0.5~3h。本发明中,所述的呱西替柳粗品为待进一步纯化的呱西替柳固体混合物。呱西替柳粗品可为市售原料或通过现有技术方法制备得到,所得的结果均一致。本发明还提供了一种呱西替柳晶型化合物的药物组合物,该药物组合物为含有呱西替柳晶体的干混悬剂。晶体的形成机理很复杂,一个新晶体的获得也具有很大的偶然性,有时不同的溶剂、在不同的结晶条件下会产生相同的晶体结构。某些特定晶型也并非一定会获得更加有利的理化性质。药物的稳定性、吸湿性、溶解性、生物活性、毒性等性质会因晶型的不同而产生巨大的差异。本申请经过大量的实验筛选研究,在实验研究过程中,本申请研究人员还进行了如下实验:(1)采用现有技术的方法用乙醇或CHCl3重结晶或采用CN103102271B的方法用甲醇、乙醇、异丙醇、正丙醇或异丁醇冷却析晶,或将甲醇、乙醇、异丙醇、正丙醇、异丁醇及CHCl3两种、三种或多种溶剂混合结晶或降温析晶或采用现有技术的工艺制备晶体。(2)采用本发明的结晶方法,分别采用不同的溶剂(乙醇或CHCl3重结晶或采用CN103102271B的方法用甲醇、乙醇、异丙醇、正丙醇或异丁醇冷却析晶,或将甲醇、乙醇、异丙醇、正丙醇、异丁醇及CHCl3两种、三种或多种溶剂,本发明采用的溶剂组合除外)制备晶体。(3)采用本发明的溶剂(氯仿和甲醇),分别采用现有技术的结晶方法制备晶体。(4)采用本发明的结晶方法和溶剂,分别改变本发明结晶方法中的降温温度、析晶过程中甲醇加入速度等,制备晶体。结果发现,均得到晶型I化合物,且其所得到的晶型I化合物均存在水溶性不好(<0.013mg/ml),有关物质含量高(>0.4%),单杂含量(>0.1%)。研究表明,在X-射线粉末衍射图谱中,由新晶型得到的衍射谱图对于特定的晶型往往是特征性的,其中谱带(尤其是在低角度)的相对强度可能会因为结晶条件、粒径和其它测定条件的差异而产生的优势取向效果而变化。因此,衍射峰的相对强度对所针对的晶型并非是特征性的,判断是否与已知的晶型相同时,更应该注意的是峰的相对位置而不是它们的相对强度。本发明经过大量的试验研究,惊喜地发现采用本发明的结晶方法条件及溶剂得到了一种与现有技术晶型I具有明显不同峰的相对位置的新的呱西替柳晶型II化合物。本发明的呱西替柳新晶型II化合物提高了水溶性且具有较高的稳定性,有关物质含量显著降低,制备方法简单易操作,制成药物组合物后用药安全性大大提高,非常适合临床应用。下面通过对本发明提供的呱西替柳晶型II化合物进行研究来解释和说明本发明技术方案:1、晶型检测取本发明制备得到的呱西替柳结晶II,使用Cu-Kα射线测量得到的X-射线粉末衍射图如图1所示,其以2θ±0.2衍射角表示的X-射线粉末衍射图在9.16°、9.53°、10.84°、11.50°、12.84°、13.79°、14.32°、14.79°、15.47°、16.26°、18.45°、18.92°、19.65°、20.54°、21.53°、21.83°、22.56°、23.12°、23.79°、25.44°、26.55°、27.36°和28.46°处显示有特征峰。2、差热分析及热重分析对本发明制备的呱西替柳晶体进行差热和热重分析,结果如附图2所示;结果表明,本品在92℃处有吸热峰。本品经熔点测定:90~93℃,从侧面证明了其为一种不同的晶型。3、水分分析采用卡式水分测定仪测定,本发明的呱西替柳结晶的含水量为0.01%。4、纯度检测经HPLC纯度检测,本发明制备得到的呱西替柳结晶的纯度可达到99.97%-99.99%。与现有技术相比,本发明具有如下优点:(1)本发明所提供的呱西替柳晶型II化合物是一种不同于现有技术的新晶型;(2)本发明所提供的呱西替柳晶型II化合物具有较好的水溶性和较高的稳定性,有关物质含量显著降低,制备方法简单易操作,制成药物组合物后用药安全性大大提高,非常适合临床应用。附图说明图1为本发明制备的呱西替柳晶型II化合物的X-射线粉末衍射图谱;图2本发明制备的呱西替柳晶型II化合物的TG-DSC图谱。具体实施例以下用实施例对本发明的技术方案进行详细说明,将有助于对本发明的技术方案的优点、效果有更进一步的了解,实施例不限定本发明的保护范围,本发明的保护范围由权利要求来决定。实施例1:呱西替柳晶型化合物的制备取呱西替柳100g于反应瓶中,加入1000ml氯仿和甲醇的混合溶液(氯仿和甲醇的体积比为3:1),加热搅拌溶解;边搅拌,边降温至-5℃(降温幅度为每10分钟5℃),向溶液中按1.0mL/min的流速加入甲醇2000ml至出晶,继续降温至-5℃(降温幅度为每10分钟1℃),保温搅拌养晶至析晶完全。真空抽滤,滤饼于40℃真空干燥6h,得91.4g白色固体,收率91.4%。所制得晶体的使用Cu-Kα射线测量得到的X-射线粉末衍射谱图如图1所示。实施例2:呱西替柳晶型化合物的制备取呱西替柳100g于反应瓶中,加入1500ml氯仿和甲醇的混合溶液(氯仿和甲醇的体积比为2:1),加热搅拌溶解;边搅拌,边降温至-3℃(降温幅度为每10分钟1℃),向溶液中按1.5mL/min的流速加入甲醇3000ml至出晶,继续降温至-10℃(降温幅度为每10分钟1℃),保温搅拌养晶至析晶完全。真空抽滤,滤饼于40℃真空干燥4h,得90.1g白色固体,收率90.1%。所制得晶体的使用Cu-Kα射线测量得到的X-射线粉末衍射谱图与实施例1相似。实施例3:呱西替柳晶型化合物的制备取呱西替柳150g于反应瓶中,加入1500ml氯仿和甲醇的混合溶液(氯仿和甲醇的体积比为2:1),加热搅拌溶解;边搅拌,边降温至0℃(降温幅度为每10分钟5℃),向溶液中按2mL/min的流速加入甲醇3000ml至出晶,继续降温至-5℃(降温幅度为每10分钟1℃),保温搅拌养晶至析晶完全。真空抽滤,滤饼于40℃真空干燥5h,得137.3g白色固体,收率91.5%。所制得的呱西替柳晶体使用Cu-Kα射线测量得到的X-射线粉末衍射谱图与实施例1相似。实施例4:呱西替柳晶型化合物的制备取呱西替柳80g于反应瓶中,加入800ml氯仿和甲醇的混合溶液(氯仿和甲醇的体积比为3:1),加热搅拌溶解;边搅拌,边降温至0℃(降温幅度为每10分钟3℃),向溶液中按2mL/min的流速加入甲醇4000ml至出晶,继续降温至-5℃(降温幅度为每10分钟1℃),保温搅拌养晶至析晶完全。真空抽滤,滤饼于40℃真空干燥5h,得74g白色固体,收率92.4%。所制得的呱西替柳晶体使用Cu-Kα射线测量得到的X-射线粉末衍射谱图与实施例1相似。实施例5:呱西替柳晶型化合物的制备取呱西替柳80g于反应瓶中,加入1600ml氯仿和甲醇的混合溶液(氯仿和甲醇的体积比为2:1),加热搅拌溶解;边搅拌,边降温至0℃(降温幅度为每10分钟5℃),向溶液中按2mL/min的流速加入甲醇3200ml至出晶,继续降温至-5℃(降温幅度为每10分钟1℃),保温搅拌养晶至析晶完全。真空抽滤,滤饼于40℃真空干燥5h,得72.6g白色固体,收率90.8%。所制得的呱西替柳晶体使用Cu-Kα射线测量得到的X-射线粉末衍射谱图与实施例1相似。下面通过实验例进一步说明本发明:实验例1:晶型化合物制备试验条件筛选表1试验条件筛选结果实验例2:流动性实验本实验例采用固定漏斗法测定各实施例样品的休止角,从而评价本发明提供的呱西替柳结晶的流动性。具体方法如下:将漏斗置于坐标纸上的适宜高度,取实施例1-5批制备的样品,从固定的漏斗中自由留下,直到形成的圆锥体顶部与漏斗口接触,测算物料堆积层的斜边与水平线的夹角度数(休止角θ)。实验结果如表2所示。表2:流动性实验结果样品12345平均值θ(°)31.231.431.031.231.231.2从表2的实验结果分析,本发明实施例1-5制备得到的呱西替柳结晶的流动性很好,有利于提高分装的准确性,并且与其他成分混合时易于混合均匀。实验例3:溶解度测定试验品:本发明实施例1-3所制备的晶型II;对照品1是市售呱西替柳原料药;对照品2是参照文献《超声波作用下合成药物呱西替柳》医药农药,2009年第02期,陈小全,仇玉芹等,制备的呱西替柳晶型I;对照品3是参照文献《呱西替柳的催化合成》武汉化工学院学报,2001年第23卷第4期,陈立钦,白崇荣,高锁,制备的呱西替柳晶型I;对照品4是参照文献《呱西替柳的合成》中国医药工业杂志,1990年21卷第9期,孙长安,黄建军,制备的呱西替柳晶型I;对照品5是参照文献《呱西替柳的合成工艺改进》华西药学杂志,1998年13卷第3期,张大永,徐鸣夏,制备的呱西替柳晶型I;对照品6是参照文献《呱西替柳的合成工艺改进》王秋芬,郑更修等,制备的呱西替柳晶型I;对照品7是参照文献《呱西替柳合成方法的改进》黑龙江大学自然科学学报,1993年第10卷第1期,赵书清,邢有权等,制备的呱西替柳晶型I;对照品8是参照文献《呱西替柳合成方法的改进》海军医高专学报,1997年第19卷第3期,祁铁流,薛荣,韦玮,制备的呱西替柳晶型I;对照品9是参照专利CN104744261A,一种呱西替柳的生产方法,张云,制备的呱西替柳晶型I;对照品10是参照文献《醋柳愈酯的相转移催化合成》天津化工,2001年第1期,何明华,林家逊,制备的呱西替柳晶型I;对照品11是参照专利103102271B制备的呱西替柳晶型I。参照中国药典2015年版二部凡例测定其溶解性,方法:取本品适量,分别加入水,每隔5分钟强力振摇30秒钟,观察30分钟内的溶解情况,即得,结果见表3。表3本发明的晶型和对照品在水中溶解性试验结果将上述实施例1-3溶解的水溶液样品在25℃恒温搅拌72小时,取样5ml。样品经0.45μm微孔滤膜过滤,弃去初滤液,取续滤液20μL测定药物含量即为水中溶解度(mg/ml)。结果见表4:表4本发明晶型与现有技术晶型在水中的溶解度对比从表3-4可以看出,25℃下,本发明呱西替柳晶型II化合物的在水中的溶解度与现有技术相比,有显著提高。实验例4:有关物质检测本实验例对实施例1~5所制备的呱西替柳结晶中的有关物质杂质进行检测分析,按照中国药典2015版第二部附录ⅪⅩF药品杂质分析指导原则进行,结果见表5。表5:各实施例样品杂质检测分析结果样品总杂(%)最大单杂(%)实施例10.080.009实施例20.090.010实施例30.090.009实施例40.080.008实施例50.070.009实验例5、稳定性试验本实验例通过加速试验和长期试验,考察本发明提供的呱西替柳晶型II化合物的稳定性。1、加速试验取实施例1-3制备的样品,于温度40±2℃、相对湿度75±5%的条件下放置6个月,分别于0、1、2、3、6个月末取样测定性状、有关物质、最大单杂,结果见表6。表6:加速试验结果(温度40±2℃,相对湿度75±5%)2、长期试验取实施例1-3制备的样品,于温度25±2℃、相对湿度60±5%的条件下放置6个月,分别于0、3、6、9、12、18、24个月末取样测定性状、有关物质、最大单杂,结果见表7。表7:长期试验结果(温度25±2℃,相对湿度60±5%)呱西替柳各杂质结构中均含有水杨酸(或乙酰水杨酸)和愈创木酚结构单元,其分解产物均为低毒性物质,直接影响药品质量和用药安全。因此,有效地降低杂质含量,哪怕是零点几个百分点,也可以有效地降低不良反应的发生,因此杂质含量对药品质量和人民用药安全至关重要。药品从生产到流通过程中需要贮存和运输才能治病救人,因此,贮存和运输过程中药品的质量显得尤为重要,稳定性是决定药品质量好坏的关键,在药品贮存和运输过程中,稳定性不好,杂质变化大直接影响人民用药安全。从表5-7可以看出,本发明呱西替柳晶型II化合物的有关物质、最大单杂含量均很低,且在温度40±2℃、相对湿度75±5%的条件下放置6个月,有关物质含量没有明显升高,各指标均无明显变化;在温度25±2℃、相对湿度60±5%的条件下放置24个月稳定,各指标均无明显变化;有效地提升了药品安全性及存储的稳定性,降低了不良反应的发生。对本发明其它实施例的呱西替柳晶型II化合物也进行了上述试验,其获得的结果相似。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1