共轭聚合物和含酯键的共轭聚合物及其制备方法和应用与流程
本发明涉及蛋白组装领域,具体涉及一种共轭聚合物,一种共轭聚合物的制备方法,一种含酯键的共轭聚合物,一种含酯键的共轭聚合物的制备方法,一种抑制和/或破坏蛋白质组装的方法以及一种抑制蛋白寡聚体形成的方法。
背景技术:
蛋白质间的相互作用是各项生物功能的基础。然而一旦蛋白质发生错误的折叠和组装,不仅会导致其失去正常的生物活性,还会引起多种棘手的疾病。组织中淀粉样多肽和蛋白的形成在大概四十种神经退行性疾病和系统性疾病中扮演重要角色,例如阿尔兹海默症、帕金森症、亨廷顿氏舞蹈症、ⅱ型糖尿病等,对人类的健康造成了严重的威胁。
过去针对调控淀粉样蛋白组装的研究主要集中在通过天然产物小分子、主客体、分子镊子、人工分子伴侣等亲疏水作用来实现。然而这种相互作用力较弱,而且易受环境因素的影响。研究出更加有效的方法来抑制和破坏淀粉样蛋白的组装仍然是一个挑战。
技术实现要素:
本发明的目的是为了克服现有技术存在的调控淀粉样蛋白组装的效果不稳定的缺陷,提供了一种含酯键的共轭组合物及其制备方法和应用,采用本发明的含酯键的共轭聚合物,能够抑制蛋白质的组装和寡聚,还可以破坏已经形成的蛋白质组装体。
具体地,第一方面,本发明提供了一种共轭聚合物,该共轭聚合物的结构如式(i)所示:
其中,r1、r2、r3和r4各自独立地为h或c1-c5的烷基且r1、r2、r3和r4中至少一个为h;n1、n2、n3和n4各自独立地为3-12的整数,n为10-14的整数。
第二方面,本发明还提供了一种共轭聚合物的制备方法,所述制备方法包括:在催化剂和碱性物质存在的条件下,将结构如式(ii)所示的化合物与结构如式(iii)所示的化合物接触,
其中,r1、r2、r3和r4各自独立地为h或c1-c5的烷基且r1、r2、r3和r4中至少一个为h;x1和x2为卤素,n1、n2、n3和n4各自独立地为3-12的整数。
第三方面,本发明提供了一种含酯键的共轭聚合物,所述含酯键的共轭聚合物的结构如式(iv)所示,
其中,r5、r6和r7各自独立地为h、c1-c5的烷基或
第四方面,本发明还提供了一种含酯键的共轭聚合物的制备方法,该制备方法包括:在碱性物质存在下,将结构如式(i)所示的共轭聚合物与4-硝基苯基氯甲酸酯接触。
第五方面,本发明还提供了一种抑制和/或破坏蛋白质组装的方法,该方法包括:将结构如式(iv)所示的含酯键的共轭聚合物与样品进行接触,所述样品为含有蛋白质组装体的样品和/或能够形成而未形成蛋白质组装体的样品。
第六方面,本发明还提供了一种抑制蛋白寡聚体形成的方法,该方法包括:将结构如式(iv)所示的含酯键的共轭聚合物与能够形成蛋白寡聚体而未形成的蛋白质单体进行接触。
本发明提供的含酯键的共轭聚合物能够蛋白质发生共价相互作用,利用化学发应使聚合物和蛋白质共价结合以产生高效的抑制效果,上述方法不仅可以抑制蛋白质的组装,还可以破坏已经形成的组装体。进一步的,本发明还可以抑制具有更高毒性的蛋白质寡聚体的形成,明显的降低由蛋白质组装引起的细胞毒性,为解决淀粉样沉积相关的疾病提供了重要思路。
附图说明
图1为实施例3中不同浓度aβ42与ppv-np作用的荧光光谱图;
图2为实施例3中不同反应时间aβ42与ppv-np作用的荧光光谱图;
图3为实施例3中klvff和ppv-np反应后的核磁图;
图4为实施例4中蛋白和ppv-np或ppv-oh在相互作用前后的圆二色光谱图;
图5为实施例4中aβ42组装后的形貌图;
图6为实施例5中aβ42组装体和ppv-np作用后的圆二色谱图;
图7为实施例6中aβ42寡聚体的斑点印迹图;
图8为实施例7中经不同处理的pc12细胞存活率图;
图9为实施例8中ppv-np对amylin和calcitonin组装的抑制图。
具体实施方式
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
本发明提供了一种共轭聚合物,该共轭聚合物的结构如式(i)所示:
其中,r1、r2、r3和r4各自独立地为h或c1-c5的烷基且r1、r2、r3和r4中至少一个为h;n1、n2、n3和n4各自独立地为3-12的整数,n为6-16的整数。
优选地,r1、r2、r3和r4各自独立地为h或c1-c3的烷基,n1、n2、n3和n4各自独立地为4-10的整数,更优选为4-8的整数;n为10-14的整数。
优选地,r1和r2均为h,r3和r4均为甲基,n1和n2为8,n3和n4为4,所述共轭聚合物的结构如下所示:
本发明还提供了一种共轭聚合物的制备方法,该制备方法包括:在催化剂和碱性物质存在的条件下,将结构如式(ii)所示的化合物与结构如式(iii)所示的化合物接触,
其中,r1、r2、r3和r4各自独立地为h或c1-c5的烷基且r1、r2、r3和r4中至少一个为h;x1和x2为卤素,n1、n2、n3和n4各自独立地为3-12的整数。
优选地,r1、r2、r3和r4各自独立地为h或c1-c3的烷基,x1和x2为i,n1、n2、n3和n4各自独立地为4-10的整数,更优选为4-8的整数。
优选地,r1和r2均为h,r3和r4均为甲基,n1和n2为8,n3和n4为4。
在本发明中,所述结构如式(ii)所示的化合物、结构如式(iii)所示的化合物、催化剂和碱性物质的用量比例可以在较大范围内变动。例如,结构如式(ii)所示的化合物、结构如式(iii)所示的化合物、催化剂和碱性物质的摩尔比可以为1:(0.8-1.2):(0.08-0.12):(2-3),优选为1:(0.9-1.1):(0.09-0.11)(2.2-2.8)。
在本发明中,所述结构如式(ii)所示的化合物和结构如式(iii)所示的化合物可以通过商购获得,或者通过本领域常规的化学合成的方法获得。例如,以r1和r2均为h、x1和x2均为i、n1和n2为8为例,结构如式(ii)所述的化合物的制备方法可以包括:在惰性气氛(零族气体或氮气)下,将2,5-二碘-1,4-苯二酚与23-羟基-3,6,9,12,15,18,21-七氧杂二十三烷4-甲基苯磺酸酯进行接触,接触的条件可以包括:温度为50-70℃,时间为25-40h。当r1和r2、x1和x2、n1和n2为本发明范围内的其他种类或数值时,可以参照上述方法进行变化,具体变化的方式为本领域技术人员所熟知。同理,以r3和r4均为甲基、n3和n4为4为例说明结构如式(iii)所示的化合物的制备方法:在惰性气氛(零族气体或氮气)下,将13,13'-((2,5-二碘-1,4-亚苯基)双氧基)双(2,5,8,11-四氧杂十三烷)与三丁基乙烯基锡以及pd(pph3)4进行接触;上述接触的条件可以包括:温度为90-120℃,时间为12-20h。
本领域技术人员应该理解的是,本发明所述的所有制备方法还可以包括对所得产物进行提纯的步骤,对于提纯的方法没有特别的要求,可以采用本领域技术人员常规使用的各种提纯方法,例如,可以采用萃取剂萃取,干燥剂干燥,并通过柱层析等方法除杂。
在本发明中,所述催化剂包括主催化剂和催化剂配体,所述主催化剂可以为醋酸钯和/或pd(pph3)4,所述催化剂配体可以为三(邻甲基苯基)磷;所述碱性物质为三正丁胺和/或三乙胺。
在本发明中,所述结构如式(ii)所示的化合物与结构如式(iii)所示的化合物接触的条件没有特别的限定,例如,所述接触条件可以包括:温度为85-120℃,优选为90-110℃,时间为40-60h,优选为45-55h。
本发明还提供了一种含酯键的共轭聚合物,所述含酯键的共轭聚合物的结构如式(iv)所示,
其中,r5、r6和r7各自独立地为h、c1-c5的烷基或
优选地,r5、r6和r7各自独立地为h或c1-c3的烷基,n1、n2、n3和n4各自独立地为4-10的整数,更优选为4-8的整数;n为10-14的整数;
优选地,r5为h,r6和r7均为甲基,n1和n2为8,n3和n4为4。
本发明还提供了一种含酯键的共轭聚合物的制备方法,,该制备方法包括:在碱性物质存在下,将结构如式(i)所示的共轭聚合物与4-硝基苯基氯甲酸酯接触。结构如式(i)所示的共轭聚合物可以按照前述方法制得,因此,含酯键的共轭聚合物的制备方法还可以包括按照上述方法制备结构如式(i)所示的化合物的步骤。
在本发明中,结构如式(i)所示的共轭聚合物、4-硝基苯基氯甲酸酯和碱性物质的用量比例可以在较大范围内变动,例如,结构如式(i)所示的化合物、4-硝基苯基氯甲酸酯和碱性物质的摩尔比可以为1:(3-8):(2-7),优选为1:(4-7):(3-6)。
在本发明中,所述结构如式(i)所示的共轭聚合物与4-硝基苯基氯甲酸酯接触的条件没有特别的限定,例如,所述接触的条件可以包括:温度为-10至40℃,优选为-5至35℃;时间为20-40h,优选为20-30h。
在本发明中,所述碱性物质可以为三正丁胺和/或三乙胺,优选为三乙胺。可以与制备结构如式(i)所示的化合物的方法中使用的碱性物质相同或不同。
本发明还提供了一种抑制和/或破坏蛋白质组装的方法,该方法包括:将结构如式(iv)所示的含酯键的共轭聚合物与样品进行接触,所述样品为含有蛋白质组装体的样品和/或能够形成而未形成蛋白质组装体的样品。结构如式(iv)所示的含酯键的共轭聚合物在上文中已有描述,在此不再赘述。
术语“蛋白质组装体”指的是蛋白质聚集形成的寡聚物或纤维,术语“蛋白质单体”指的是单分散形式存在的蛋白。蛋白质单体组装形成蛋白质组装体的条件可以根据蛋白的种类进行确定,例如β-淀粉样蛋白(aβ)、胰岛淀粉样多肽(amylin)以及降钙素(calcitonin)在37℃组装,胰岛素在65℃组装。
根据本发明,所述样品为能够形成而未形成蛋白质组装体的样品时,所述接触的条件包括:温度为37±0.5℃,时间为10min以上,优选为5-30h。根据本发明的这种实施方式,所述能够形成蛋白质组装体的蛋白质单体可以为本领域各种能够形成蛋白质组装体的蛋白质单体,例如可以为β-淀粉样蛋白(aβ)、胰岛淀粉样多肽(amylin)、降钙素(calcitonin)、胰岛素、tau蛋白和α-突触核蛋白中的一种或多种。优选地,样品中的蛋白质单体与结构如式(i)所示的共轭聚合物的摩尔比可以为1:0.01-20,优选为1:0.1-10。
在本发明中,所述样品为含有蛋白质组装体的样品时,所述接触的条件可以包括:温度为37±0.5℃,时间为10min以上,优选为1-10h。根据本发明的这种实施方式,所述能够形成蛋白质组装体的蛋白质单体可以为本领域各种能够形成蛋白质组装体的蛋白质单体,例如可以为β-淀粉样蛋白(aβ)、胰岛淀粉样多肽(amylin)、降钙素(calcitonin)、胰岛素、tau蛋白和α-突触核蛋白中的一种或多种。上述蛋白可以商购获得。所述蛋白质单体形成蛋白质组装体的条件可以根据蛋白的种类进行确定。
优选地,样品中的以蛋白质单体计的蛋白质组装体与含酯键的共轭聚合物的摩尔比为1:0.01-20,优选为1:0.1-10。所述以蛋白质单体计的蛋白质组装体指的是按照蛋白质单体的投料量计算蛋白质组装体的量。
由于本发明的含酯键的共轭聚合物能够抑制和/或破坏蛋白的组装,故能够降低蛋白组装体对细胞的毒性。
本发明还提供了一种抑制蛋白寡聚体形成的方法,该方法包括:将结构如式(iv)所示的含酯键的共轭聚合物与能够形成而未形成蛋白寡聚体的蛋白质单体进行接触。
术语“蛋白质寡聚体”指的是以低聚体为主,一种低相对分子质量的蛋白聚集体。所述蛋白质寡聚体的形成条件可以根据蛋白质的种类进行确定。
在本发明中,结构如式(iv)所示的含酯键的共轭聚合物与能够形成蛋白寡聚体而未形成的蛋白质单体进行接触的条件包括:温度可以为20-30℃,优选为22-28℃;时间可以为8-20h,优选为10-15h。
根据本发明,所述能够形成蛋白质寡聚体的蛋白质单体可以为本领域各种能够形成蛋白质寡聚体的蛋白质单体,例如可以为β-淀粉样蛋白(aβ)、胰岛淀粉样多肽(amylin)、降钙素(calcitonin)、胰岛素、tau蛋白和α-突触核蛋白中的一种或多种。上述蛋白可以商购获得。
根据一种具体的实施方式,商购获得的蛋白可以使用六氟异丙醇进行预处理使蛋白质以单体的形式的存在,β-淀粉样蛋白(aβ)、胰岛淀粉样多肽(amylin)、降钙素(calcitonin)均可以使用六氟异丙醇预处理的方式,其他蛋白可以根据蛋白的具体种类和性质使其保持单体的存在形式,可以为本领域的常规选择。
在本发明中,样品中的以蛋白质单体计的蛋白质寡聚体与含酯键的共轭聚合物的摩尔比可以为1:0.01-20,优选为1:0.1-10。所述以蛋白质单体计的蛋白质寡聚体指的是按照蛋白质单体的投料量计算蛋白质寡聚体的量。
蛋白质组装体和蛋白寡聚体会引起细胞毒性,因此,通过抑制和/或破坏蛋白质组装、抑制蛋白寡聚体形成可有效避免神经细胞损伤,提高细胞存活率。
以下将通过实施例对本发明进行详细描述。
以下实施例中,aβ42购自lifetechnologies公司,货号为74335783a;amylin和calcitonin购自苏州强耀公司;寡聚物多克隆抗体a11购自lifetechnologies公司,货号为qd215255;aβ单克隆抗体6e10购自covance公司,货号为803001;二抗hrp购自中杉金桥公司,货号为zb-2301;催化剂pd(pph3)4购自sigma-aldrich公司,货号为216666;催化剂配体p(o-tol)3购自sigma-aldrich公司,货号为287822;其余试剂均为常规市售产品,为分析纯或色谱纯;pc12细胞(atcccrl-1721);
荧光检测在hitachif-4500仪器上进行;圆二色光谱检测在hitachijasco-j815仪器上进行;透射电镜(tem)购自hitachis-7700;
室温为25℃。
实施例1
本实施例用于说明结构如式(i)所示的共轭聚合物的制备方法
(1)单体的制备
r1和r2均为h,r3和r4均为甲基,n1和n2为8,n3和n4为4,式(ii)
所示单体的为单体3,式(iii)所示的单体为单体5:
单体3的制备:在10ml丙酮中加入k2co3(0.23g,1.68mmol),通入n230min以除去溶剂中的o2。在n2保护下加入化合物2,5-二碘-1,4-苯二酚(0.20g,0.56mmol)和23-羟基-3,6,9,12,15,18,21-七氧杂二十三烷4-甲基苯磺酸酯(0.59g,1.12mmol),将反应温度升至60℃,在n2气氛下反应30h。冷却至室温后,过滤洗涤沉淀。收集丙酮溶液用无水na2so4干燥,过滤,旋蒸除去溶剂。粗产物用硅胶柱层析分离纯化,洗脱剂为石油醚/乙酸乙酯/甲醇(1/1/1,v/v),得淡黄色液体(0.13g,22%)。1hnmr(400mhz,dmso-d6,δ):7.37(s,1h),4.55(t,j=4.0hz,1h),4.09(t,j=6.0hz,2h),3.74(t,j=4.0hz,2h),3.62(m,2h),3.50(m,24h),3.42(m,2h).13cnmr(400mhz,cdcl3,δ):153.01,123.36,87.35,72.79,70.61,70.32,70.23,70.17,69.41,60.66.hr-ms(maldi,[m+k]):calcd.forc38h68i2ko18:1105.2127;found:1105.2132.
单体5的制备:将13,13'-((2,5-二碘-1,4-亚苯基)双氧基)双(2,5,8,11-四氧杂十三烷)(0.50g,0.67mmol)溶解在12ml甲苯中,通入n230min以除去溶剂中的o2,在n2保护下向该溶液中加入三丁基乙烯基锡(1.27g,4mmol)和乙醇洗涤过的pd(pph3)4(16mg),升温至100℃c反应16h。停止加热待反应液冷却至室温后,用40ml浓度为2m的kf水溶液除去未反应的三丁基乙烯基锡。收集有机相,用无水na2so4干燥,过滤,旋蒸除去溶剂。粗产物用硅胶柱层析分离纯化,洗脱剂为石油醚/乙酸乙酯/甲醇(1/1/1,v/v),得无色油状液体(0.21g,59%)。1hnmr(300mhz,cdcl3,δ):7.07-6.98(m,4h),5.74(dd,j=16.5,1.2hz,2h),5.26(dd,j=9.9,1.2hz,2h),4.13(t,j=4.8hz,4h),3.85(t,j=5.4hz,4h),3.75-3.63(m,20h),3.53(m,4h),3.37(s,6h).13cnmr(400mhz,cdcl3,δ):151.00,131.68,127.91,114.61,111.54,72.24,71.16,70.98,70.95,70.93,70.82,70.18,69.40,59.30.hr-ms(maldi,[m+k]):calcd.forc28h46ko10:581.2723.;found:581.2728.
(2)共轭聚合物ppv-oh(结构如下所示)的制备
在n2保护下将单体3(0.21g,0.20mmol)、单体5(0.11g,0.20mmol),催化剂醋酸钯(10mg,0.013mmol),催化剂配体p(o-tol)3(20mg,0.066mmol)和三正丁胺(119μl,0.50mmol)溶解在5ml的dmf中,通n230min以除去溶剂中氧气,反应液升温至100℃,在氮气保护下反应48h。停止加热待反应液冷却至室温后,用甲醇透析2天,期间更换甲醇10次,透析袋截留分子量为7500,最后将甲醇旋蒸除去,得到深红色油状液体(0.18g,56%)。1hnmr(400mhz,dmso-d6,δ):7.52(br,2h),7.25(br,2h),4.53(br,1h),4.21-3.39(br,48h),3.19(s,3h)。gpc:mw=29020,pdi=1.15。
实施例2
本实施例用于说明含酯键的共轭聚合物ppv-np的制备
将ppv-oh(15mg,0.01mmol)溶于2ml超干二氯甲烷中,加入三乙胺(5.06mg,0.05mmol),在0℃搅拌下滴加4-硝基苯基氯甲酸酯(12.09mg,0.06mmol)的二氯甲烷溶液。在0℃下继续反应1h后移至室温继续反应24h。加入10ml二氯甲烷稀释,用饱和食盐水洗三次,用无水na2so4干燥后,浓缩,在乙醚中沉降,得到深红色油状液体(12.3mg,74%)。1hnmr(400mhz,dmso-d6,δ):8.30(br,1h),7.54(br,3h),7.25(br,2h),4.34-4.05(br,5h),3.86(br,4h),3.67-3.36(br,38h),3.18(s,3h)。由核磁数据分析4-硝基苯基甲酸酯的取代度约为50%。
实施例3
本实施例用于说明含酯键的共轭聚合物ppv-np和蛋白的相互作用
(1)蛋白的准备
将冻干的aβ42、amylin、calcitonin溶解在六氟异丙醇中,配成1mg/ml。超声10min后在4℃震荡2h以进一步溶解。随后,将溶液分装在离心管中,氮吹除去溶剂,真空干燥2h。于-20℃储存。
(2)通过监测ppv-np和aβ42作用后的荧光变化来研究它们之间的相互作用。
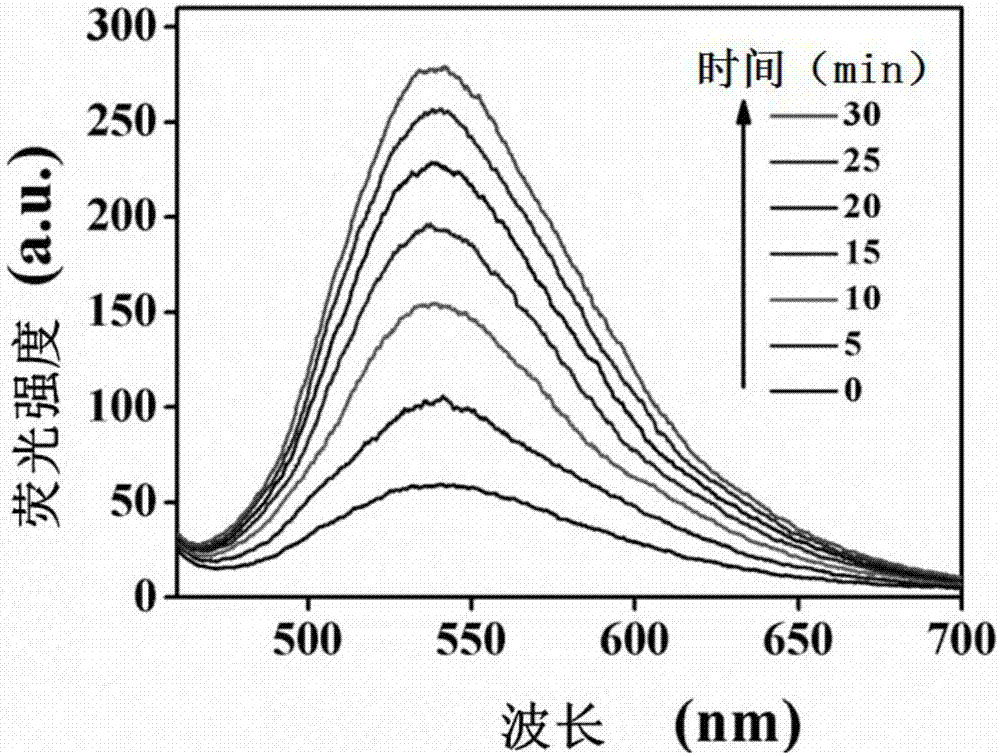
在浓度分别为0、5、10、20、30、40、60、80μm的aβ42溶液中加入ppv-np,使ppv-np的终浓度为10μm,37℃搅拌反应30min,用荧光光谱仪表征不同浓度蛋白下ppv-np的荧光变化。在37℃下将终浓度为80μm的aβ42和10μm的ppv-np混合,监测不同作用时间后ppv-np的荧光变化。结果分别如图1和图2所示。
图1为不同浓度aβ42与ppv-np作用的荧光光谱图。从图1中可以看出,随着aβ42浓度的增加,ppv-np的荧光逐渐增强。
图2为不同反应时间aβ42与ppv-np作用的荧光光谱图。从图2中可以看出,随着反应时间的延长,ppv-np的荧光逐渐增强。
(3)通过核磁谱图变化研究ppv-np和aβ42的相互作用。
将klvffae(aβ42的核心序列,作为模型来研究核磁,2.0mm)和ppv-np(4.0mm)在室温下搅拌反应过夜,溶剂为dmso-d6。
图3为klvffae和ppv-np反应后的核磁图。从图3中可以看出明显的酰胺质子化学位移变化,表明klvffae和ppv-np之间存在疏水相互作用。在高场处赖氨酸侧链ε位亚甲基化学位移的变化表明共价反应的发生。
实施例4
本实施例用于说明本发明提供的抑制蛋白组装的方法
将六氟异丙醇处理后aβ42溶于10mm的naoh中,4℃离心(14000g,10min),取上清液稀释在10mm的pb溶液中,aβ42的终浓度为40μm。为了进一步验证ppv-np对aβ42组装的共价抑制作用,选用不含反应性基团的ppv-oh做阴性对照。将aβ42分别与ppv-np、ppv-oh在1:0,1:1,1:2,1:5,1:10摩尔比下37℃搅拌作用30min后,将上述溶液移至透析管透析3h,透析液为10mm的naoh(总体积的10%)与10mm的pb溶液(总体积的90%)。透析后在缓慢的搅拌下继续37℃培养15h。取200μl样品用圆二色光谱检测aβ42组装后的二级结构变化,光路长为1mm,扫描波长190-260nm,扫描速度为100nm/min,响应时间为4s,每个光谱是重复扫描3次的平均值。用tem进一步表征ppv-np对aβ42组装的抑制效果,取5μl培养后样品,滴到普通碳膜铜网上,作用2min后,用超纯水洗两次,用3重量%的醋酸双氧铀染色1min后,用滤纸吸走染色液。晾干后,用tem观察。
图4为相互作用前后的圆二色光谱图。从图4中可以看出单独的aβ42培养后在216nm处有负峰,说明aβ42是以β折叠的形式存在的,而随着加入ppv-np量的不断增加,在216nm处的负峰逐渐变弱至消失,在198nm处出现新的负峰,表明aβ42以无规卷曲的构象存在,即ppv-np抑制了aβ42的组装。加入了ppv-oh的阴性对照组在培养后在216nm左右有负峰,和单独培养aβ42相似,说明不含反应性基团的ppv-oh不能抑制aβ42的组装。
图5为aβ42组装后的形貌。从图5中可以看出,单独的aβ42和加入ppv-oh的样品组有纤维形成,加入ppv-np的样品没有形成纤维组装体。
实施例5
本实施例用于说明本发明提供的破坏蛋白质组装的方法
采用与实施例4相同的方法单独培养aβ42,使其形成aβ42组装体。aβ42组装体与10倍摩尔过量的ppv-np在37℃下搅拌反应2h,透析3h除去dmso,用圆二色谱检测aβ42的构象。对照组为不加ppv-np在相同操作下加等量dmso。
图6为aβ42组装体和ppv-np作用后的圆二色谱图。从图6中可以看出,加入ppv-np后aβ42恢复为无规卷曲构象,说明ppv-np可以破坏已经形成的组装体。
实施例6
本实施例用于说明本发明提供的抑制蛋白寡聚体形成的方法
取六氟异丙醇处理后的aβ42,加dmso溶解成2mm,加入10mm的pb溶液稀释成40μm,然后分别与ppv-np、ppv-oh、dmso作用30min,其中ppv-np、ppv-oh为10倍摩尔过量,dmso的体积和ppv-np体积相同。随后,将样品移入透析管,于室温下透析12h,透析液为10mm的pb溶液,透析后将各个样品定容为相同的体积。
利用斑点印迹法检测aβ42寡聚体的形成。在硝酸纤维膜上分别滴2μl样品,风干。用含10重量%脱脂牛奶的10mm的tbst(含tween0.01体积%)室温震荡1h后,用tbst溶液洗三次,每次5分钟。将寡聚物多克隆抗体a11以1:500(体积)或aβ单克隆抗体6e10以1:1000(体积)稀释到含3重量%bsa的tbst中,在摇床上室温作用一小时,用tbst溶液洗三次,每次5分钟。然后分别用相应的二抗hrp孵育,利用增强的化学发光法检测寡聚体的形成。
图7为aβ42寡聚体的斑点印迹图。从图7中可以看出,ppv-np可以抑制aβ42寡聚体的形成。
实施例7
本实施例用于说明ppv-np对aβ42组装体引起细胞毒性的抑制
pc12细胞在含10体积%牛血清的r1640培养基中置于37℃含5%二氧化碳的恒温培养箱培养。pc12细胞接种在96孔板内,密度为8×103个/孔,放入37℃培养箱过夜贴壁培养。含有ppv-np和ppv-oh以及单独的aβ42在37℃培养16h后,用培养基稀释至浓度为20μm后加入到96孔板中与细胞作用48h。用mtt法对pc12细胞存活分析,去掉96孔板中上清液,加入mtt(100μl/孔)在37℃培养4小时,去掉上清液,加入dmso(100μl/孔),将96孔板放入酶标仪中测570nm处的吸收。
图8为经不同处理的pc12细胞存活率图。由图8可知单独的aβ42和加入ppv-oh组对细胞有明显的毒性,细胞的存活率约为46%。而加入了ppv-np后可以明显的降低对pc12的细胞毒性,细胞的存活率约为100%。
实施例8
本实施例用于说明ppv-np抑制蛋白amylin或calcitonin组装的方法
取六氟异丙醇处理后的amylin,加入10mm的naoh(总体积的10%),用10mm的pb溶液(总体积的90%)稀释至40μm。分别加入十倍摩尔过量的ppv-np、ppv-oh及与聚合物等体积的dmso。37℃下搅拌反应30min,移至透析管培养4h,透析液组成和缓冲液相同。取样品进行圆二色谱测定和tem表征。
取六氟异丙醇处理后的calcitonin,加dmso溶解成3.9mm,用10mm的pb溶液(含100mm的nacl)稀释至60μm。分别加入十倍摩尔过量的ppv-np、ppv-oh及与聚合物等体积的dmso。37℃下搅拌反应30min,用透析管透析3h后转移至微量反应管中继续搅拌培养8h。取样品进行圆二色谱测定和tem表征。
图9为ppv-np对amylin和calcitonin(简称ct)组装的抑制。从圆二色谱图中可以看出加入ppv-np后,amylin和calcitonin仍然以无规卷曲构象存在,而单独的amylinn、calcitonin和加入ppv-oh的主要以β折叠的形式存在。tem也进一步表明ppv-np可以有效抑制amylin和calcitonin组装。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!