一种硅基碳硼烷聚合物及其制备方法与流程
本发明涉及一种硅基碳硼烷聚合物及其制备方法,属于精细化学品和耐高温聚合物合成技术领域。
技术背景
sibcn陶瓷作为一种新型高性能陶瓷材料,比sic、si3n4、bn等二元体系陶瓷和sicn、sico等三元体系的陶瓷具有更优异的高温稳定性和抗氧化性,有些sibcn陶瓷的高温稳定性甚至可达到2000℃。正因如此,sibcn陶瓷数十年以来作为陶瓷涂层、陶瓷纤维、陶瓷基复合材料的研究方兴未艾。此外还可用作陶瓷的粘接剂、电脑芯片的多层连接等,因此被广泛应用于信息、电子、航空航天和军事等领域。
sibcn陶瓷主要采用有机无机杂化聚合物聚硼硅氮烷(pbsz)经高温裂解获得。其中,聚硼硅氮烷的合成主要采用两类合成路径,分别为单体途径和聚合物途径。
单体途径的特征是先利用含有硼或者硅的单元反应生成分别含有硼、硅的单体,然后再聚合成sibcn陶瓷前驱体,但副产物难去除,无法工业化。聚合物路径的特征是利用含硅聚合物与含硼化合物反应,制得陶瓷前驱体,但聚合物途径在前驱体制备收率及后续陶瓷化产率方面均明显低于单体途径,因此成本较高;或者先将多乙烯基环硅氮烷硼烷化,利用环硅氮烷的反应性交联获得陶瓷体,然而,将硅氮烷进行硼烷化的方法,由于使用的有机硼烷具有剧毒,生产受到限制,可操作性差,难以工业化实施。
技术实现要素:
本发明针对现有的聚硼硅氮烷陶瓷前驱体制备工艺或复杂或采用有毒原料,以及合成成本高等不足,提供了一种新型的新型硅基碳硼烷聚合物及其制备方法。所公开的嵌段聚合物由无毒的碳硼烷合成制得,由于特有的合成路线得到的网状结构,拥有更高的陶瓷化产率,并且可由市售材料在非常温和的条件下制得,适合放大生产,便于在工业中推广应用。
本发明公开了一种硅基碳硼烷聚合物,其化学结构式如下:
其中,r=-ch3或-ph;m、n分别为3~50的整数。
本发明还公开了上述的硅基碳硼烷聚合物的制备方法,包括如下步骤:
(1)以1,1′-烃基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷为原料,在碱性条件下、金属催化剂下,进行反应制备1,1′-烃基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷;
(2)以二甲基氯硅烷、三乙烯基三甲基环三硅氮烷为原料,在金属催化剂下,反应制备1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷;
(3)以1,1′-烃基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷、1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷为原料,在三乙胺存在下,制备所述硅基碳硼烷聚合物。
上述技术方案中,所述1,1′-烃基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷为1,1′-甲基苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷或1,1′-双苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷;所述金属催化剂为钯碳催化剂或铂碳催化剂;所述碱性条件为氢氧化钠、一水磷酸二氢钠条件。
上述技术方案中,步骤(1)反应完成后,过滤反应液,然后滤液经萃取、水洗、干燥、除溶剂得到1,1′-烃基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷;步骤(3)反应完成后,过滤反应液,然后滤液经萃取、水洗、干燥、除溶剂得到所述硅基碳硼烷聚合物。
上述技术方案中,
步骤(1)为,将氢氧化钠、一水磷酸二氢钠与水混合制备缓冲液,再加入金属催化剂、1,1′-烃基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷,反应制备1,1′-烃基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷;
步骤(2)为,惰性气氛下,在三乙烯基三甲基环三硅氮烷溶液中加入金属催化剂;然后加入二甲基氯硅烷溶液;反应制备1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷;
步骤(3)为,将含有三乙胺的1,1′-烃基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷滴加入1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷中,反应制备所述硅基碳硼烷聚合物。
上述技术方案中,
步骤(1)为,将氢氧化钠与水混合,再加入一水磷酸二氢钠混合制备缓冲液,再加入醚类溶剂以及金属催化剂;然后加入1,1′-烃基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷的芳烃溶液,反应制备1,1′-烃基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷;
步骤(2)为,惰性气氛下,在三乙烯基三甲基环三硅氮烷芳烃溶液中加入金属催化剂;然后加入二甲基氯硅烷芳烃溶液;反应制备1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷;
步骤(3)为,将含有三乙胺的1,1′-烃基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷芳烃溶液滴加入1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷中,反应制备所述硅基碳硼烷聚合物。
上述技术方案中,所述醚类溶剂为1,4-二氧六环,所述芳烃为甲苯和/或二甲苯;所述惰性气氛为氮气气氛。
上述技术方案中,
步骤(1)中,氢氧化钠、水、一水磷酸二氢钠、金属催化剂、1,1′-烃基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷的质量比为(0.2~2)∶(20~200)∶(0.2~5)∶(0.1~2)∶(0.5~5);
步骤(2)中,三乙烯基三甲基环三硅氮烷、金属催化剂、二甲基氯硅烷的质量比为(0.5~5)∶(0.001~0.2)∶(0.5~5);
步骤(3)中,三乙胺、1,1′-烃基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷的质量比为(0.5~10)∶(1~5)。
上述技术方案中,
步骤(1)中,反应温度为25℃~65℃,反应时间为1~48小时;
步骤(2)中,反应温度为25℃~85℃,反应时间为1~48小时;
步骤(3)中,反应温度为25℃~75℃,反应时间为1~48小时。
本发明还公开了1,1′-烃基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷的制备,惰性气体下,向烃基二氯硅烷溶液内滴加乙炔基溴化镁格氏试剂,于35~45℃下反应;然后滴加癸硼烷溶液,于80~90℃下反应,得到烃基硅甲撑双碳硼烷;惰性气体下,冰水浴条件下,向烃基硅甲撑双碳硼烷溶液内滴加正丁基锂溶液,冰水浴条件下反应;然后滴加二甲基氯硅烷溶液,于冰水浴条件下下反应,然后于室温反应,得到1,1′-烃基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷。烃基二氯硅烷、乙炔基溴化镁格氏试剂、癸硼烷的质量比为(0.2~2)∶(0.5~5)∶(0.1~1);烃基硅甲撑双碳硼烷、正丁基锂、二甲基氯硅烷的质量比为(1~10)∶(0.2~2)∶(0.5~5)。
本发明还公开了一种sibcn陶瓷的制备方法,将上述的硅基碳硼烷聚合物经过成型处理得到sibcn陶瓷。通过现有方式实现硅基碳硼烷聚合物的成型处理,比如载体涂覆后风干,或者进一步热处理等。
本发明还公开了一种sibcn陶瓷,由上述的硅基碳硼烷聚合物经过成型处理得到。
本发明具体反应式如下:
其中,r=-ch3或-ph;m、n分别为3~50的整数。
本发明公开的新型硅基碳硼烷聚合物,其以工业化原料三乙烯基三甲基环三硅氮烷为骨架支撑,通过与无毒的碳硼烷单体聚合反应引入耐热单元,得到的预聚物以其四元体系和网状交联结构可以提供非常优异的耐热性能和抗氧化性能,其简易的合成路线以及温和的反应条件,便于推广应用于制备sibcn陶瓷材料。
制备硅基碳硼烷聚合物的具体操作步骤为:
(1)
a.将氢氧化钠,去离子水和一水磷酸二氢钠混合均匀,制备成缓冲溶液。
b.将缓冲溶液加入到反应器中,再加入1,4-二氧六环以及贵金属催化剂,然后加入1,1′-烃基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷溶于芳烃溶剂中所形成的溶液,进行羟基化反应。
c.反应结束后,过滤除去不溶的贵金属催化剂,滤液加入萃取剂萃取。
d.合并萃取液,用去离子水洗至中性,经干燥、旋蒸,得到1,1′-烃基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷。
(2)
a.由二甲基氯硅烷溶解于除水芳烃溶剂中,配制二甲基氯硅烷溶液,备用。
b.在反应器中投入三乙烯基三甲基环三硅氮烷和除水芳烃溶剂,氮气保护下,加入铂催化剂和上述a中配制好的二甲基氯硅烷溶液,在氮气保护下保温反应一定时间,得到1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷溶液。
(3)
a.将1,1′-烃基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷溶解于除水芳烃溶剂中,加入三乙胺,配制成碳硼烷单体溶液,备用。
b.在氮气保护下,将碳硼烷单体溶液滴加到上述(2)中制备的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷溶液中进行缩聚反应,反应温度保持在25℃~75℃,反应时间为1~48小时。
c.反应结束后,等待反应液自然冷却至室温,过滤除去不溶的三乙胺盐酸盐,滤液加入萃取剂萃取,每次加入萃取剂5~30份,共萃取3~5次。
d.合并萃取液,然后用去离子水水洗至中性,加入0.5~20份干燥剂干燥,旋蒸除去萃取剂,得到产物硅基碳硼烷聚合物。
比如上述的硅基碳硼烷聚合物的制备方法,可以如下
(1)
a.按重量计,将0.2~2份氢氧化钠,20~200份去离子水,混合均匀,再加入0.2~5份一水磷酸二氢钠,搅拌溶解,制备成缓冲溶液,备用。
b.按重量计,将上述a中配制好的缓冲溶液加入到反应器中,再加入5~50份的1,4-二氧六环以及0.1~2份的贵金属催化剂,最后加入0.5~5份1,1′-烃基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷溶于10~60份芳烃溶剂中所形成的溶液,在温度为25℃~65℃的条件下,进行羟基化反应,反应时间为1~48小时。
c.反应结束后,过滤除去不溶的贵金属催化剂,滤液加入萃取剂萃取,每次加入萃取剂5~30份,共萃取3~5次。
d.合并萃取液,然后用去离子水洗至中性,加入1~20份干燥剂干燥30分钟~8小时,旋蒸除去萃取剂,得到产物,1,1′-烃基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷。
(2)
a.按重量计,由0.5~5份二甲基氯硅烷,5~50份除水芳烃溶剂配制的二甲基氯硅烷溶液,备用。
b.按重量计,在反应器中投入0.5~5份三乙烯基三甲基环三硅氮烷和5~50份除水芳烃溶剂,氮气保护下,加入0.001~0.2份铂催化剂,再加入上述a中配制好的二甲基氯硅烷溶液。设定反应温度为25~85℃,在氮气保护下搅拌反应,反应时间为1~48小时,得到1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷。
(3)
a.按重量计,将1~5份1,1′-烃基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷溶解于1~50份除水芳烃溶剂中,再加入0.5~10份三乙胺。在氮气保护下,将该溶液滴加到上述(2)中制备的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷溶液中进行缩聚反应,反应温度保持在25℃~75℃,反应时间为1~48小时。
b.反应结束后,等待反应液自然冷却至室温,过滤除去不溶的三乙胺盐酸盐颗粒,滤液加入萃取剂萃取,每次加入萃取剂5~30份,共萃取3~5次。
合并萃取液,然后用去离子水水洗至中性,加入0.5~20份干燥剂干燥,旋蒸除去萃取剂,得到产物硅基碳硼烷聚合物。
本发明中,所述的萃取剂为乙醚,乙酸乙酯,正己烷或其中任意两者以任意重量比混合的溶剂;所述的干燥剂为无水硫酸镁,无水硫酸钠,无水氯化钙中的任意一种;所述的芳烃溶剂为甲苯,二甲苯或两者以任意比例混合的混合溶剂;所述的旋蒸除萃取剂的条件为温度30~60℃,真空度为10~20mmhg。
本发明与现有技术相比的突出优点是:
1.与现有技术中含硅碳硼烷聚合物不同,本发明公开的硅基碳硼烷聚合物分子主链上含环状三硅氮烷单元,该结构单元可赋予预聚物以充分的交联反应性和交联程度,有利于后续交联反应。
2.本发明公开的硅基碳硼烷聚合物由工业化原料环状三硅氮烷与无毒的碳硼烷交替共聚而成,其中的环状三硅氮烷具有三个取代反应官能团,由此得到其与二官能度的碳硼烷交替共聚形成交联网络结构,该交联网络结构非常均匀,这也有利于硅氮元素、硼元素分布更均匀,有利于聚合物作为耐高温材料的性能发挥。
3.本发明公开的新型硅基碳硼烷聚合物分子结构中具有双碳硼烷夹心结构,硼元素含量更高,从而拥有更好的耐热性能和耐热氧化性能。
4.本发明制备新型硅基碳硼烷聚合物所使用的原材料均为市售原料,无毒安全,来源广泛,价格便宜。制备反应条件温和、工艺简单且产物易于提纯,适用工业化生产。
附图说明
图1是实施例一制备的1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷的红外吸收曲线图;
图2是实施例一制备的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷的红外吸收曲线图;
图3是实施例一制备的新型硅基碳硼烷聚合物的红外吸收曲线图;
图4是实施例一制备的新型硅基碳硼烷聚合物的氢核磁共振光谱图;
图5是实施例一、实施例二、实施例三、实施例四中涂层的碳纤维以及未涂层的碳纤维的质量残留与烘烤时间的关系图。
具体实施方式
下面结合附图和实施例对本发明技术方案作进一步的阐述。
实施例一
1.合成1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷
在1000ml的三口烧瓶中加入135克除水四氢呋喃和13.2克的二苯基二氯硅烷,搭好实验装置后,先通氮气除去装置内的空气,而后通过恒压滴液漏斗滴加25克的乙炔基溴化镁格氏试剂,滴加时间为30分钟,设置反应温度为40℃,反应时间为4小时,同时配置6.2克癸硼烷,78克乙腈和178克四氢呋喃的混合溶液,反应4小时之后,同样通过恒压滴液漏斗滴加,滴加时间为30分钟。滴加结束之后,调节反应温度为80℃,反应时间为48小时。
反应结束后,加入配置好的110克乙腈,55克丙酮,36克浓盐酸和50克去离子水混合溶液进行淬灭,淬灭时间为6小时,直至不再有气泡产生。淬灭结束后,加入无水乙醚对反应液进行萃取,每次加入200克乙醚,共萃取3次。合并萃取液,分出油层,然后用去离子水洗至中性,加入30克无水硫酸镁干燥5小时,最后过滤,于45℃、真空度为20mmhg旋蒸至恒重,得到9.8g双苯基硅甲撑双碳硼烷。
在100ml三口烧瓶中加入17.82克干燥四氢呋喃和2.36克双苯基硅甲撑双碳硼烷,搭好反应装置后,先通氮气除去装置内的空气,在冰浴的条件下等三口烧瓶中溶液的温度降至0℃,而后通过恒压滴液漏斗滴加6.00克1.6m正丁基锂己烷溶液,滴加时间为15分钟,反应时间为2小时,保持反应温度为零下。反应两个小时后,同样在冰浴的条件下滴加0.95克二甲基氯硅烷和8.92克干燥四氢呋喃的混合溶液,滴加过程为15分钟,反应1小时后转移至室温的条件下继续反应,反应时间为24小时。
反应结束后,加入20.58克饱和氯化铵溶液进行淬灭,淬灭时间为12小时。淬灭结束后,用无水乙醚对反应液进行萃取,分液,然后用去离子水洗至中性,4.00克无水硫酸镁干燥5小时,最后过滤,于35℃、真空度为20mmhg下旋蒸至恒重,得到产物1,1′-双苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷。
配缓冲溶液:将0.20克氢氧化钠加入到50.00g去离子水中配置成0.1mol/l的氢氧化钠溶液,溶解后加入0.65克的一水磷酸二氢钠,搅拌溶解。
在250ml的三口烧瓶中先加入缓冲溶液,15.51克的1,4-二氧六环和0.50克的钯碳催化剂,搭好实验装置后,通过恒压滴液漏斗滴加配置好的3.00克1,1′-双苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷和17.50克除水甲苯的混合溶液,滴加时间为5分钟,设置反应温度为35℃,反应时间为12小时。
反应结束后,抽滤除去不溶的钯碳催化剂,滤液用无水乙醚萃取,每次加入20克无水乙醚,共萃取3次。合并萃取液,分出油层,然后用去离子水水洗至中性,加入5克无水硫酸镁干燥5小时,最后过滤,于55℃,真空度为20mmhg旋蒸至恒重,得到产物1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷
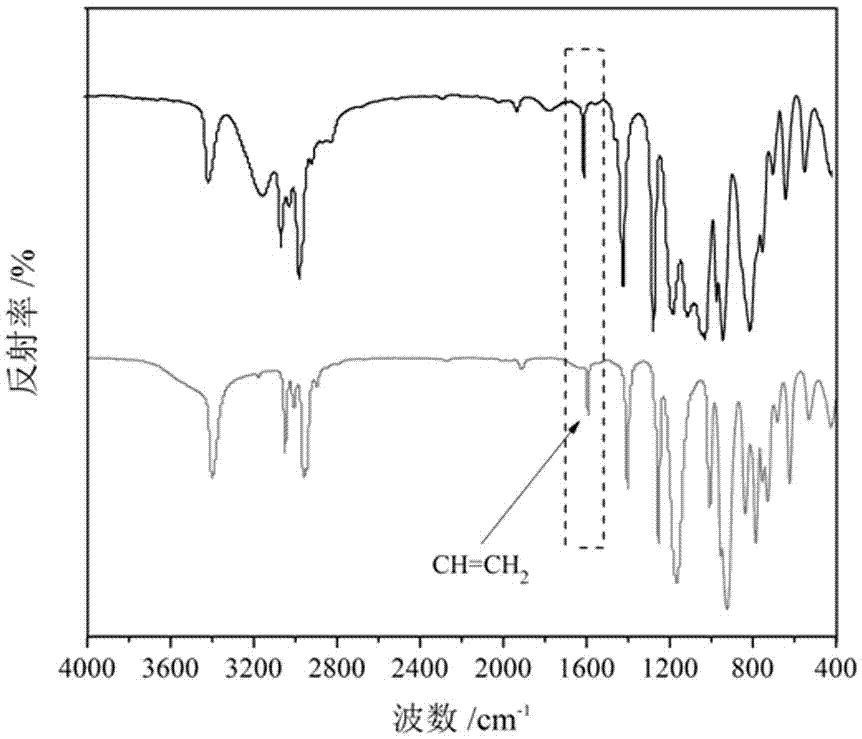
在100ml的三口烧瓶中先加入17.50克除水甲苯溶液,1.27克的三乙烯基三甲基环三硅氮烷,以及0.01克卡斯特催化剂,搭好实验装置后,先通氮气除去装置内的空气,而后通过恒压滴液漏斗滴加配置好的1.42克二甲基氯硅烷和8.75克除水甲苯的混合溶液,滴加时间为5分钟,在25℃的条件下反应1小时,随后升温至75℃反应4小时,反应液变为银灰色,得到产物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷,直接进入下一步反应。
3.缩聚反应
在100ml的烧杯中加入13.00克的无水甲苯,3.07克1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷,以及1.02克的三乙胺,混合均匀,备用。
在上述步骤2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷中,通过恒压滴液漏斗滴加配制好的碳硼烷单体溶液,滴加时间为15分钟,在室温下反应30分钟后,升温至50℃,反应时间为18小时。
反应结束后,自然冷却至室温,抽滤除去不溶的三乙胺盐酸盐,滤液用25ml无水乙醚萃取3次,分液,然后用去离子水水洗至中性,5克无水硫酸镁干燥5小时,最后过滤,在55℃、真空度为20mmhg下旋蒸至恒重,得到产物,一种新型的硅基碳硼烷聚合物。
参见附图1,是按本实施例技术方案制备的1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷的红外吸收曲线图谱。从图中可以看到,2129.13cm-1处为si-h的伸缩振动峰消失以及843.15cm-1处为si-oh的伸缩振动峰的出现,可以有效的证明了硅羟基化反应的进行。
参见附图2,是按本实施例技术方案制备的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷的红外吸收曲线图谱。从图中可以看到,1594.83cm-1处为c=ch2的伸缩振动峰减弱,还留有部分的c=ch2峰是由于反应不完全,但也足以证明了硅氢加成反应的发生。
参见附图3,是按本实施例技术方案制备的新型硅基碳硼烷聚合物的红外吸收曲线图谱。从图中可以看到,3379.83cm-1处的吸收峰是n-h的伸缩振动峰;碳硼烷的b-h峰在2563.46cm-1处;而843.15cm-1处为si-oh的伸缩振动峰的消失以及1031.58cm-1处si-o-sicm-1伸缩振动峰的出现则有力的证实了缩聚反应的发生。
参见附图4,是按本实施例技术方案制备的新型硅基碳硼烷聚合物氢核磁共振光谱。从图中可以看到,0.05-0.26(m,si-ch3);0.4-2.0(br,b-h);7.2-7.7(d,ph-h);2.27(d,n-h);5.86-6.08(m,ch=ch2);结合红外吸收图谱,证实了聚合物的合成。
产物分子结构式如下:
式中,m和n分别4~8的整数
4.涂层处理
将硅基碳硼烷聚合物溶解在二氯甲烷溶液中,配制成质量浓度为0.12g/毫升的溶液,备用。将t-300碳纤维在氮气保护和400℃的条件下烘烤2小时,再在质量浓度为5%的硝酸溶液中浸泡1小时,然后去离子水淋洗至中性。再将处理好的碳纤维浸泡在硅基碳硼烷聚合物的二氯甲烷溶液中,室温自然风干。
重复处理3次,得到表面有3层sibcn陶瓷涂层的碳纤维,碳纤维的拉伸强度为3982mpa。
测试了处理前后碳纤维的耐热稳定性能,结果见附图5。从图中可以看出,未经处理的碳纤维在500℃的条件下,80分钟便已经完全降解,而经过3次涂层处理的碳纤维,即使加热至900℃的条件下烘烤2小时后,质量残留高达99%。
实施例二
1.合成1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷
在1000ml的三口烧瓶中加入135克除水四氢呋喃和13.2克的二苯基二氯硅烷,搭好实验装置后,先通氮气除去装置内的空气,而后通过恒压滴液漏斗滴加25克的乙炔基溴化镁格氏试剂,滴加时间为30分钟,设置反应温度为40℃,反应时间为4小时,同时配置6.2克癸硼烷,78克乙腈和178克四氢呋喃的混合溶液,反应4小时之后,同样通过恒压滴液漏斗滴加,滴加时间为30分钟。滴加结束之后,调节反应温度为86℃,反应时间为48小时。
反应结束后,加入配置好的110克乙腈,55克丙酮,36克浓盐酸和50克去离子水混合溶液进行淬灭,淬灭时间为6小时,直至不再有气泡产生。淬灭结束后,加入无水乙醚对反应液进行萃取,每次加入200克乙醚,共萃取3次。合并萃取液,分出油层,然后用去离子水洗至中性,加入30克无水硫酸镁干燥5小时,最后过滤,于45℃、真空度为20mmhg旋蒸至恒重,得到9.8g双苯基硅甲撑双碳硼烷。
在250ml三口烧瓶中加入35.64克干燥四氢呋喃和4.75克双苯基硅甲撑双碳硼烷,搭好实验装置后,先通氮气除去装置内的空气,在冰浴的条件下等三口烧瓶中溶液的温度降至0℃而后通过恒压滴液漏斗滴加12.50克1.6m正丁基锂己烷溶液,滴加时间为20分钟,反应时间为4小时,保持反应温度为零下。反应4个小时后,同样在冰浴的条件下滴加1.92克二甲基氯硅烷和17.85克干燥四氢呋喃的混合溶液,滴加过程为20分钟,反应1小时后转移至室温的条件下继续反应,反应时间为24小时。
反应结束后,加入41.16克饱和氯化铵溶液进行淬灭,淬灭时间为20小时。淬灭结束后,用无水乙醚对反应液进行萃取,分液,然后用去离子水洗至中性,10克无水硫酸镁干燥5小时,最后过滤,于35℃、真空度为20mmhg下旋蒸至恒重,得到产物1,1′-双苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷。
配置缓冲溶液:将0.40克氢氧化钠加入到100.00g去离子水中配置成0.10mol/l的氢氧化钠溶液,溶解后加入1.30克的一水磷酸二氢钠,搅拌溶解。
在250ml的三口烧瓶中先加入缓冲溶液,32.00克1,4-二氧六环和1.50克的钯碳催化剂,搭好实验装置后,通过恒压滴液漏斗滴加配置好的6.00克1,1′-双苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷和35.50克除水甲苯的混合溶液,滴加时间为5分钟,设置反应温度为35℃,反应时间为12小时。
反应结束后,抽滤除去不溶的钯碳催化剂,滤液用无水乙醚萃取,每次加入40克无水乙醚,共萃取5次。合并萃取液,分出油层,然后用去离子水水洗至中性,加入15克无水硫酸镁干燥5小时,最后过滤,于60℃,真空度为20mmhg旋蒸至恒重,得到产物1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷
在250ml的三口烧瓶中先加入35.00克除水甲苯溶液,2.54克的三乙烯基三甲基环三硅氮烷,以及0.02克卡斯特催化剂,搭好实验装置后,先通氮气除去装置内的空气,而后通过恒压滴液漏斗滴加配置好的2.84克二甲基氯硅烷和17.75克除水甲苯的混合溶液,滴加时间为10分钟,在35℃的条件下反应1小时,随后升温至75℃反应4小时,反应液变为银灰色,得到产物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷,直接进入下一步反应。
3.缩聚反应
在250ml的烧杯中加入26.00克的无水甲苯,6.54克1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷,以及2.04克的三乙胺,混合均匀,备用。
在上述步骤2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷中,通过恒压滴液漏斗滴加配制好的碳硼烷单体溶液,滴加时间为30分钟,在室温下反应1小时后,升温至55℃,反应时间为24小时。
反应结束后,自然冷却至室温,抽滤除去不溶的三乙胺盐酸盐,滤液用无水乙醚萃取,分液,然后用去离子水水洗至中性,10克无水硫酸镁干燥5小时,最后过滤,在55℃、真空度为15mmhg下旋蒸至恒重,得到产物硅基碳硼烷聚合物。
m和n分别为5~10的整数
4.涂层处理
将硅基碳硼烷聚合物溶解在二氯甲烷溶液中,配制成质量浓度为0.36g/毫升的溶液,备用。
将t-300碳纤维在氮气保护和400℃的条件下烘烤2小时,再在质量浓度为5%的硝酸溶液中浸泡1小时,然后去离子水淋洗至中性。再将碳纤维浸泡在硅基碳硼烷聚合物的二氯甲烷溶液中,室温自然风干。重复处理3次得到表面有3层涂层的碳纤维,测试了处理前后碳纤维的耐热稳定性能,结果见附图5。加热至700℃的条件下烘烤2小时,测得处理后碳纤维质量残余70.39%。
实施例三
1.合成1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷
合成双苯基硅甲撑双碳硼烷同实施例一。
在1000ml三口烧瓶中加入71.28克干燥四氢呋喃和9.55克双苯基硅甲撑双碳硼烷,搭好实验装置后,先通氮气除去装置内的空气,在冰浴的条件下等三口烧瓶中溶液的温度降至0℃而后通过恒压滴液漏斗滴加25.50克1.6m正丁基锂己烷溶液,滴加时间为60分钟,反应时间为8小时,保持反应温度为零下。反应8个小时后,同样在冰浴的条件下滴加3.95克二甲基氯硅烷和35.75克干燥四氢呋喃的混合溶液,滴加过程为30分钟,反应2小时后转移至室温的条件下继续反应,反应时间为24小时。
反应结束后,加入82.32克饱和氯化铵溶液进行淬灭,淬灭时间为20小时。淬灭结束后,用无水乙醚对反应液进行萃取,分液,然后用去离子水洗至中性,20克无水硫酸镁干燥5小时,最后过滤,于40℃、真空度为20mmhg下旋蒸至恒重,得到产物1,1′-双苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷。
配制缓冲溶液:将0.80克氢氧化钠加入到200.00g去离子水中配置成0.10mol/l的氢氧化钠溶液,溶解后加入2.60克的一水磷酸二氢钠,搅拌溶解。
在250ml的三口烧瓶中先加入缓冲溶液,64.00克1,4-二氧六环和3.50克的钯碳催化剂,搭好实验装置后,通过恒压滴液漏斗滴加配置好的12.50克1,1′-双苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷和71.50克除水甲苯的混合溶液,滴加时间为30分钟,设置反应温度为55℃,反应时间为12小时。
反应结束后,抽滤除去不溶的钯碳催化剂,滤液用无水乙醚萃取,每次加入100克无水乙醚,共萃取3次。合并萃取液,分出油层,然后用去离子水水洗至中性,加入50克无水硫酸镁干燥5小时,最后过滤,于55℃,真空度为20mmhg旋蒸至恒重,得到产物1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷
在1000ml的三口烧瓶中先加入75.50克除水甲苯溶液,5.25克的三乙烯基三甲基环三硅氮烷,以及0.05克卡斯特催化剂,搭好实验装置后,先通氮气除去装置内的空气,而后通过恒压滴液漏斗滴加配置好的5.75克二甲基氯硅烷和35.50克除水甲苯的混合溶液,滴加时间为30分钟,在45℃的条件下反应1小时,随后升温至75℃反应5小时,反应液变为银灰色,得到产物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷,直接进入下一步反应。
3.缩聚反应
在1000ml的烧杯中加入55.00克的无水甲苯,6.50克1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷,以及2.50克的三乙胺,混合均匀,得到碳硼烷单体溶液,备用。
在上述步骤2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷中,通过恒压滴液漏斗滴加配制好的碳硼烷单体溶液,滴加时间为45分钟,在室温下反应120分钟后,升温至55℃,反应时间为36小时。
反应结束后,自然冷却至室温,抽滤除去不溶的三乙胺盐酸盐,滤液用无水乙醚萃取,分液,然后用去离子水水洗至中性,50克无水硫酸镁干燥12小时,最后过滤,在55℃、真空度为20mmhg下旋蒸至恒重,得到产物,一种新型的硅基碳硼烷聚合物。
产物分子结构式如下:
式中,m和n分别4~6的整数
4.涂层处理
将硅基碳硼烷聚合物溶解在二氯甲烷溶液中,配制成质量浓度为0.12g/毫升的溶液,备用。将t-300碳纤维在氮气保护和400℃的条件下烘烤2小时,再在质量浓度为5%的硝酸溶液中浸泡1小时,然后去离子水淋洗至中性。再将处理好的碳纤维浸泡在碳硼烷陶瓷前驱体的二氯甲烷溶液中,室温自然风干。重复处理3次得到表面有3层涂层的碳纤维t-300。
测试了处理前后碳纤维的耐热稳定性能,结果见附图5。将处理后碳纤维加热至800℃烘烤2小时,发现处理后碳纤维在800℃下氧化60分钟以后质量保持恒定,且碳纤维质量残余76.65%。
实施例四
1.合成1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷
在2000ml三口烧瓶中加入145.50克干燥四氢呋喃和19.32克双苯基硅甲撑双碳硼烷(实施例二),搭好实验装置后,先通氮气除去装置内的空气,在冰浴的条件下等三口烧瓶中溶液的温度降至0℃而后通过恒压滴液漏斗滴加55.25克1.6m正丁基锂己烷溶液,滴加时间为120分钟,反应时间为8小时,保持反应温度为零下。反应8个小时后,同样在冰浴的条件下滴加7.68克二甲基氯硅烷和71.45克干燥四氢呋喃的混合溶液,滴加过程为60分钟,反应2小时后转移至室温的条件下继续反应,反应时间为36小时。
反应结束后,加入164.64克饱和氯化铵溶液进行淬灭,淬灭时间为20小时。淬灭结束后,用无水乙酸乙酯对反应液进行萃取,分液,然后用去离子水洗至中性,40克无水硫酸镁干燥12小时,最后过滤,于35℃、真空度为10mmhg下旋蒸至恒重,得到产物1,1′-双苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷。
配制缓冲溶液:将1.60克氢氧化钠加入到400.00g去离子水中配置成0.10mol/l的氢氧化钠溶液,溶解后,加入5.20克的一水磷酸二氢钠,搅拌溶解。
在2000ml的三口烧瓶中先加入缓冲溶液,135.50克的1,4-二氧六环和6.50克的钯碳催化剂,搭好实验装置后,通过恒压滴液漏斗滴加配置好的24.30克1,1′-双苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷和142.50克除水甲苯的混合溶液,滴加时间为5分钟,设置反应温度为35℃,反应时间为12小时。
反应结束后,抽滤除去不溶的钯碳催化剂,滤液用无水正己烷萃取,每次加入40克无水正己烷,共萃取5次。合并萃取液,分出油层,然后用去离子水水洗至中性,加入100克无水硫酸镁干燥12小时,最后过滤,于55℃,真空度为15mmhg旋蒸至恒重,得到产物1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷
在2000ml的三口烧瓶中先加入145.00克除水甲苯溶液,10.50克的三乙烯基三甲基环三硅氮烷,以及0.10克卡斯特催化剂,搭好实验装置后,先通氮气除去装置内的空气,而后通过恒压滴液漏斗滴加配置好的11.52克二甲基氯硅烷和71.50克除水甲苯的混合溶液,滴加时间为120分钟,在35℃的条件下反应2小时,随后升温至75℃反应10小时,反应液变为银灰色,得到产物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷,直接进入下一步反应。
3.缩聚反应
在2000ml的烧杯中加入105.50克的无水甲苯,27.80克1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷,以及8.53克的三乙胺,混合均匀,备用。
在上述步骤2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷中,通过恒压滴液漏斗滴加配制好的碳硼烷单体溶液,滴加时间为120分钟,在室温下反应4小时后,升温至55℃,反应时间为48小时。
反应结束后,自然冷却至室温,抽滤除去不溶的三乙胺盐酸盐,滤液用无水正己烷萃取,分液,然后用去离子水水洗至中性,100克无水硫酸钠干燥5小时,最后过滤,在55℃、真空度为15mmhg下旋蒸至恒重,得到产物,一种新型的硅基碳硼烷聚合物。
产物分子结构式如下:
m和n分别为6~12的整数
4.涂层处理
将碳硼烷陶瓷前驱体溶解在二氯甲烷溶液中,配制成质量浓度为0.12g/毫升的溶液,备用。将碳纤维在氮气保护和400℃的条件下烘烤2小时,再在质量浓度为5%的硝酸溶液中浸泡1小时,然后去离子水淋洗至中性。再将处理好的碳纤维浸泡在碳硼烷陶瓷前驱体的二氯甲烷溶液中,室温自然风干。重复处理3次得到有3层涂层的碳纤维t-300。碳纤维加热至600℃烘烤2小时,质量残留高于80%,热稳定性优于未处理碳纤维(见附图5)。
实施例五
1.合成1,1′-甲基苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷
在1000ml的三口烧瓶中加入135克除水四氢呋喃和9.6克的甲基苯基二氯硅烷,搭好实验装置后,先通氮气除去装置内的空气,而后通过恒压滴液漏斗滴加22克的乙炔基溴化镁格氏试剂,滴加时间为35分钟,设置反应温度为40℃,反应时间为4小时,同时配置6.2克癸硼烷,78克乙腈和178克四氢呋喃的混合溶液,反应4小时之后,同样通过恒压滴液漏斗滴加,滴加时间为35分钟。滴加结束之后,调节反应温度为80℃,反应时间为48小时。
反应结束后,加入配置好的110克乙腈,55克丙酮,36克浓盐酸和50克去离子水混合溶液进行淬灭,淬灭时间为6小时,直至不再有气泡产生,淬灭结束后,用无水乙醚对反应液进行萃取,分液,然后用去离子水洗至中性,无水硫酸镁干燥5小时,最后过滤,于30℃、真空度为15mmhg旋蒸至恒重,得到产物甲基苯基硅甲撑双碳硼烷8.8g。
在100ml三口烧瓶中加入17.82克干燥四氢呋喃和2.36克甲基苯基硅甲撑双碳硼烷,搭好实验装置后,先通氮气除去装置内的空气,在冰浴的条件下等三口烧瓶中溶液的温度降至0℃,而后通过恒压滴液漏斗滴加6.00克1.6m正丁基锂己烷溶液,滴加时间为15分钟,反应时间为2小时,保持反应温度为零下。反应两个小时后,同样在冰浴的条件下滴加0.95克二甲基氯硅烷和8.92克干燥四氢呋喃的混合溶液,滴加过程为15分钟,反应1小时后转移至室温的条件下继续反应,反应时间为24小时。
反应结束后,加入20.58克饱和氯化铵溶液进行淬灭,淬灭时间为12小时。淬灭结束后,用无水乙醚对反应液进行萃取,分液,然后用去离子水洗至中性,4.00克无水硫酸镁干燥5小时,最后过滤,于35℃、真空度为20mmhg下旋蒸至恒重,得到产物1,1′-甲基苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷。
配制缓冲溶液:将0.20克氢氧化钠加入到50.00g去离子水中配置成0.1mol/l的氢氧化钠溶液,溶解后加入0.65克的一水磷酸二氢钠,搅拌溶解。
在250ml的三口烧瓶中先加入缓冲溶液,15.51克的1,4-二氧六环和0.50
克的钯碳催化剂,搭好实验装置后,通过恒压滴液漏斗滴加配置好的3.00克1,1′-甲基苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷和17.50克除水甲苯的混合溶液,滴加时间为5分钟,设置反应温度为35℃,反应时间为12小时。
反应结束后,抽滤除去不溶的钯碳催化剂,滤液用无水乙酸乙酯萃取,每次加入20克无水乙酸乙酯,共萃取3次。合并萃取液,分出油层,然后用去离子水水洗至中性,加入5克无水氯化钙干燥5小时,最后过滤,于55℃,真空度为20mmhg旋蒸至恒重,得到产物1,1′-甲基苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷
在100ml的三口烧瓶中先加入17.50克除水甲苯溶液,1.27克的三乙烯基三甲基环三硅氮烷,以及0.01克卡斯特催化剂,搭好实验装置后,先通氮气除去装置内的空气,而后通过恒压滴液漏斗滴加配置好的1.42克二甲基氯硅烷和8.75克除水甲苯的混合溶液,滴加时间为5分钟,在25℃的条件下反应1小时,随后升温至75℃反应4小时,反应液变为银灰色,得到产物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷,直接进入下一步反应。
3.缩聚反应
在100ml的烧杯中加入13.00克的无水甲苯,3.67克1,1′-甲基苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷,以及1.02克的三乙胺,混合均匀,备用。
在上述步骤2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷中,通过恒压滴液漏斗滴加配制好的碳硼烷单体溶液,滴加时间为15分钟,在室温下反应30分钟后,升温至50℃,反应时间为18小时。
反应结束后,自然冷却至室温,抽滤除去不溶的三乙胺盐酸盐,滤液用无水乙醚萃取,分液,然后用去离子水水洗至中性,5克无水硫酸镁干燥5小时,最后过滤,在55℃、真空度为20mmhg下旋蒸至恒重,得到产物,一种新型的硅基碳硼烷聚合物。
产物分子结构式如下:
m和n分别为5~12的整数
4.涂层处理
将碳硼烷陶瓷前驱体溶解在三氯甲烷溶液中,配制成质量浓度为0.12g/毫升的溶液,备用。将t-700碳纤维在氮气保护和400℃的条件下烘烤2小时,再在质量浓度为5%的硝酸溶液中浸泡1小时,然后去离子水淋洗至中性。再将处理好的碳纤维浸泡在碳硼烷陶瓷前驱体的二氯甲烷溶液中,室温自然风干。重复处理2次得到有2层涂层的碳纤维材料。碳纤维在空气的氛围中加热至1000℃质量保留率达76.24%。
实施例六
1.合成1,1′-甲基苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷
甲基苯基硅甲撑双碳硼烷同实施例五。
在250ml三口烧瓶中加入35.64克干燥四氢呋喃和4.72克甲基苯基硅甲撑双碳硼烷,搭好实验装置后,先通氮气除去装置内的空气,在冰浴的条件下等三口烧瓶中溶液的温度降至0℃,而后通过恒压滴液漏斗滴加12.50克1.6m正丁基锂己烷溶液,滴加时间为30分钟,反应时间为3小时,保持反应温度为零下。反应3个小时后,同样在冰浴的条件下滴加1.95克二甲基氯硅烷和17.85克干燥四氢呋喃的混合溶液,滴加过程为30分钟,反应2小时后转移至室温的条件下继续反应,反应时间为18小时。
反应结束后,加入41.16克饱和氯化铵溶液进行淬灭,淬灭时间为12小时。淬灭结束后,用无水乙酸乙酯对反应液进行萃取,分液,然后用去离子水洗至中性,10.00克无水硫酸镁干燥5小时,最后过滤,于40℃、真空度为20mmhg下旋蒸至恒重,得到产物1,1′-甲基苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷。
配置缓冲溶液:将0.40克氢氧化钠加入到100.00g去离子水中配置成0.1mol/l的氢氧化钠溶液,溶解后加入1.30克的一水磷酸二氢钠,搅拌溶解。
在250ml的三口烧瓶中先加入缓冲溶液,32.50克的1,4-二氧六环和1.50
克的钯碳催化剂,搭好实验装置后,通过恒压滴液漏斗滴加配置好的6.50克1,1′-甲基苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷和35.50克除水甲苯的混合溶液,滴加时间为15分钟,设置反应温度为35℃,反应时间为12小时。
反应结束后,抽滤除去不溶的钯碳催化剂,滤液用无水乙酸乙酯萃取,每次
加入20克无水乙酸乙酯,共萃取5次。合并萃取液,分出油层,然后用去离子水水洗至中性,加入10克无水氯化钙干燥5小时,最后过滤,于55℃,真空度为15mmhg旋蒸至恒重,得到产物1,1′-甲基苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷
在250ml的三口烧瓶中先加入35.50克除水甲苯溶液,2.52克的三乙烯基三甲基环三硅氮烷,以及0.04克卡斯特催化剂,搭好实验装置后,先通氮气除去装置内的空气,而后通过恒压滴液漏斗滴加配置好的2.85克二甲基氯硅烷和16.50克除水甲苯的混合溶液,滴加时间为15分钟,在35℃的条件下反应1小时,随后升温至65℃反应5小时,反应液变为银灰色,得到产物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷,直接进入下一步反应。
3.缩聚反应
在250ml的烧杯中加入26.50克的无水甲苯,6.81克1,1′-甲基苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷,以及2.42克的三乙胺,混合均匀,备用。
在上述步骤2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷中,通过恒压滴液漏斗滴加配制好的碳硼烷单体溶液,滴加时间为20分钟,在室温下反应50分钟后,升温至50℃,反应时间为24小时。
反应结束后,自然冷却至室温,抽滤除去不溶的三乙胺盐酸盐,滤液用无水正己烷萃取,分液,然后用去离子水水洗至中性,10克无水硫酸钠干燥5小时,最后过滤,在55℃、真空度为20mmhg下旋蒸至恒重,得到产物,一种新型的硅基碳硼烷聚合物。产物分子结构式如下:
m和n分别为6~12的整数
4.涂层处理
将碳硼烷陶瓷前驱体溶解在二氯乙烷溶液中,配制成质量浓度为0.12g/毫升的溶液,备用。将t900碳纤维在氮气保护和400℃的条件下烘烤2小时,再在质量浓度为5%的硝酸溶液中浸泡1小时,然后去离子水淋洗至中性。再将处理好的碳纤维浸泡在碳硼烷陶瓷前驱体的二氯甲烷溶液中,室温自然风干。重复处理3次得到有3层涂层的碳纤维材料。处理后碳纤维在空气的氛围中加热至1000℃质量保留率为75.57%。
实施例七
1.合成1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷
在2000ml三口烧瓶中加入132.68克干燥四氢呋喃和19.30克双苯基硅甲撑双碳硼烷(实施例二),搭好实验装置后,先通氮气除去装置内的空气,在冰浴的条件下等三口烧瓶中溶液的温度降至0℃而后通过恒压滴液漏斗滴加55.29克1.6m正丁基锂己烷溶液,滴加时间为120分钟,反应时间为8小时,保持反应温度为零下。反应8个小时后,同样在冰浴的条件下滴加7.67克二甲基氯硅烷和71.40克干燥四氢呋喃的混合溶液,滴加过程为60分钟,反应2小时后转移至室温的条件下继续反应,反应时间为36小时。
反应结束后,加入164.23克饱和氯化铵溶液进行淬灭,淬灭时间为20小时。淬灭结束后,用无水乙酸乙酯对反应液进行萃取,分液,然后用去离子水洗至中性,40克无水硫酸镁干燥12小时,最后过滤,于35℃、真空度为10mmhg下旋蒸至恒重,得到产物1,1′-双苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷。
配缓冲溶液:将1.60克氢氧化钠加入到400.00g去离子水中配置成0.10mol/l的氢氧化钠溶液,溶解后加入5.20克的一水磷酸二氢钠,搅拌溶解。
在2000ml的三口烧瓶中先加入缓冲溶液,135.12克的1,4-二氧六环和6.35克的钯碳催化剂,搭好实验装置后,通过恒压滴液漏斗滴加配置好的24.12克1,1′-双苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷和142.50克除水甲苯的混合溶液,滴加时间为5分钟,设置反应温度为35℃,反应时间为12小时。
反应结束后,抽滤除去不溶的钯碳催化剂,滤液用无水正己烷萃取,每次加入40克无水正己烷,共萃取5次。合并萃取液,分出油层,然后用去离子水水洗至中性,加入100克无水硫酸镁干燥12小时,最后过滤,于55℃,真空度为15mmhg旋蒸至恒重,得到产物1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷
在2000ml的三口烧瓶中先加入145克除水甲苯溶液,10.34克的三乙烯基三甲基环三硅氮烷,以及0.10克卡斯特催化剂,搭好实验装置后,先通氮气除去装置内的空气,而后通过恒压滴液漏斗滴加配置好的11.42克二甲基氯硅烷和71.50克除水甲苯的混合溶液,滴加时间为120分钟,在35℃的条件下反应2小时,随后升温至75℃反应10小时,反应液变为银灰色,得到产物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷,直接进入下一步反应。
3.缩聚反应
在2000ml的烧杯中加入105.50克的无水甲苯,24.56克1,1′-双苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷,以及8.53克的三乙胺,混合均匀,备用。
在上述步骤2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷中,通过恒压滴液漏斗滴加配制好的碳硼烷单体溶液,滴加时间为120分钟,在室温下反应4小时后,升温至55℃,反应时间为48小时。
反应结束后,自然冷却至室温,抽滤除去不溶的三乙胺盐酸盐,滤液用无水正己烷萃取,分液,然后用去离子水水洗至中性,100克无水硫酸钠干燥5小时,最后过滤,在55℃、真空度为15mmhg下旋蒸至恒重,得到产物为一种新型的碳硼烷陶瓷前驱体。
碳硼烷陶瓷前驱体分子结构式如下:
m和n分别为5~10的整数
4.涂层处理
将碳硼烷陶瓷前驱体溶解在二氯乙烷溶液中,配制成质量浓度为0.12g/毫升的溶液,备用。将碳纤维在氮气保护和400℃的条件下烘烤2小时,再在质量浓度为5%的硝酸溶液中浸泡1小时,然后去离子水淋洗至中性。再将处理好的碳纤维浸泡在碳硼烷陶瓷前驱体的二氯乙烷溶液中,室温自然风干,得到单层涂层的碳纤维t-300。碳纤维加热至600℃烘烤2小时,质量保留率为34%,热稳定性优于未处理碳纤维。涂层处理后,碳纤维的拉伸强度为4728mpa,处理后拉伸强度仅损失3.50%。
实施例八
1.合成1,1′-甲基苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷
在1000ml的三口烧瓶中加入150克除水四氢呋喃和9.6克的甲基苯基二氯硅烷,搭好实验装置后,先通氮气除去装置内的空气,而后通过恒压滴液漏斗滴加22克的乙炔基溴化镁格氏试剂,滴加时间为35分钟,设置反应温度为40°c,反应时间为4小时,同时配置6.2克癸硼烷,78克乙腈和178克四氢呋喃的混合溶液,反应4小时之后,同样通过恒压滴液漏斗滴加,滴加时间为35分钟。滴加结束之后,调节反应温度为86℃,反应时间为48小时。
反应结束后,加入配置好的110克乙腈,55克丙酮,36克浓盐酸和50克去离子水混合溶液进行淬灭,淬灭时间为6小时,直至不再有气泡产生,淬灭结束后,用无水乙醚对反应液进行萃取,分液,然后用去离子水洗至中性,无水硫酸镁干燥5小时,最后过滤,于30℃、真空度为15mmhg旋蒸至恒重,得到产物甲基苯基硅甲撑双碳硼烷8.8g。
在100ml三口烧瓶中加入17.82克干燥四氢呋喃和2.36克甲基苯基硅甲撑双碳硼烷,搭好实验装置后,先通氮气除去装置内的空气,在冰浴的条件下等三口烧瓶中溶液的温度降至0℃,而后通过恒压滴液漏斗滴加6.00克1.6m正丁基锂己烷溶液,滴加时间为15分钟,反应时间为2小时,保持反应温度为零下。反应两个小时后,同样在冰浴的条件下滴加0.95克二甲基氯硅烷和8.92克干燥四氢呋喃的混合溶液,滴加过程为15分钟,反应1小时后转移至室温的条件下继续反应,反应时间为24小时。
反应结束后,加入20.58克饱和氯化铵溶液进行淬灭,淬灭时间为12小时。淬灭结束后,用无水乙醚对反应液进行萃取,分液,然后用去离子水洗至中性,4.00克无水硫酸镁干燥5小时,最后过滤,于35℃、真空度为20mmhg下旋蒸至恒重,得到产物1,1′-甲基苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷。
配制缓冲溶液:将0.20克氢氧化钠加入到50.00g去离子水中配置成0.1mol/l的氢氧化钠溶液,溶解后加入0.65克的一水磷酸二氢钠,搅拌溶解。
在250ml的三口烧瓶中先加入缓冲溶液,15.51克的1,4-二氧六环和0.50克的钯碳催化剂,搭好实验装置后,通过恒压滴液漏斗滴加配置好的3.00克1,1′-甲基苯基硅甲撑-2,2′-双(二甲基硅基)双碳硼烷和17.50克除水甲苯的混合溶液,滴加时间为5分钟,设置反应温度为35℃,反应时间为12小时。
反应结束后,抽滤除去不溶的钯碳催化剂,滤液用无水乙酸乙酯萃取,每次加入20克无水乙酸乙酯,共萃取3次。合并萃取液,分出油层,然后用去离子水水洗至中性,加入5克无水氯化钙干燥5小时,最后过滤,于55℃,真空度为20mmhg旋蒸至恒重,得到产物1,1′-甲基苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷。
2.合成1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷
在100ml的三口烧瓶中先加入17.35克除水甲苯溶液,1.35克的三乙烯基三甲基环三硅氮烷,以及0.01克卡斯特催化剂,搭好实验装置后,先通氮气除去装置内的空气,而后通过恒压滴液漏斗滴加配置好的1.48克二甲基氯硅烷和8.88克除水甲苯的混合溶液,滴加时间为5分钟,在25℃的条件下反应1小时,随后升温至75℃反应4小时,反应液变为银灰色,得到产物1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷,直接进入下一步反应。
3.缩聚反应
在100ml的烧杯中加入13.00克的无水甲苯,3.72克1,1′-甲基苯基硅甲撑-2,2′-双(二甲基羟基硅基)双碳硼烷,以及1.05克的三乙胺,混合均匀,备用。
在上述步骤2得到的1,3,5-三甲基-1,3,5-三[β-(二甲基氯硅基)乙基]环三硅氮烷中,通过恒压滴液漏斗滴加配制好的碳硼烷单体溶液,滴加时间为15分钟,在室温下反应30分钟后,升温至50℃,反应时间为18小时。
反应结束后,自然冷却至室温,抽滤除去不溶的三乙胺盐酸盐,滤液用无水乙醚萃取,分液,然后用去离子水水洗至中性,5克无水硫酸镁干燥5小时,最后过滤,在55℃、真空度为20mmhg下旋蒸至恒重,得到产物,一种新型的硅基碳硼烷聚合物。产物分子结构式如下:
m和n分别为5~12的整数
4.涂层处理
将碳硼烷陶瓷前驱体溶解在二氯乙烷中,配制成质量浓度为0.12g/毫升的溶液,备用。将t-700碳纤维在氮气保护和400℃的条件下烘烤2小时,再在质量浓度为5%的硝酸溶液中浸泡1小时,然后以去离子水淋洗至中性。再将处理好的碳纤维浸泡在碳硼烷陶瓷前驱体的二氯乙烷溶液中,室温自然风干。重复处理5次得到有5层涂层的碳纤维材料。碳纤维在空气的氛围中加热至1000℃质量保留率达92.15%,碳纤维处理后耐热性能非常优异。
- 还没有人留言评论。精彩留言会获得点赞!