一种含烯丙基聚酰亚胺二胺单体及其聚酰亚胺聚合物与制备方法与流程
本发明属于聚酰亚胺材料合成技术领域。
背景技术:
聚酰亚胺的综合性能优异,其中包括优异的化学稳定性、良好的机械性能、耐热性能、耐辐射性能以及电性能等,已广泛应用于航天、军事、电子等领域。但由于大多数聚酰亚胺分子具有刚性的主链结构以及分子链的紧密堆积,导致了其具有较差的溶解性、熔融性和加工性能,应用领域受到了很大的限制。目前在聚酰亚胺中引入的交联封端基团主要有马来酰亚胺基团、降冰片烯二甲亚胺以及炔基封端基团,用马来酰亚胺封端的聚合物通常分子量较低,交联后的样品很脆,很难单独使用,一般都用做复合材料的基体;而用降冰片烯二甲亚胺封端的聚合物交联后通常得到的是脂肪链结构,热稳定性同芳香族相比较差;炔基封端基团因为综合改善了以上两种封端基团的缺点,成为当前在聚酰亚胺中普遍引入的交联基团,一般都用做复合材料的基体。如中国专利cn101139441a用烯丙基基团封端,聚合物分子链变短,烯丙基含量增大,但分子量偏低,从而其热力学性能和机械性能在一定程度上有所损失。中国专利cn106366335a在一定程度上改进了交联剂的类型和反应条件(如反应物浓度,反应温度等),公开一种以均苯三甲酰肼(btch)为交联剂,在50℃的条件下与pi混匀后可直接涂覆成交联的聚酰亚胺膜,其杨氏模量最高达1.56gpa,拉伸强度达66.3mpa。
但是目前这类材料由于在普通溶剂中不可溶,因此材料的加工性能较差的缺点限制了这类材料的应用范围。
技术实现要素:
本发明为了解决上述问题,提供了一类全新结构的聚酰亚胺聚合物,同时也提供了其均聚物和共聚物的制备方法,将可交联的烯丙基引入到分子主链中,通过调整烯丙基的比例来制备交联度可控的聚酰亚胺材料。因烯丙基易于在加热或紫外辐照条件下发生交联反应,具有固化时无小分子逸出、不易产生空隙等优点,并且制备方法简单易操作,制备的材料的耐热性、机械性能良好。本发明在分子主链和侧链上引入三氟甲基基团,使其在普通溶剂可溶,材料的加工性能大大增加,可满足材料在不同环境的使用要求,拓宽了这类高性能聚合物材料的应用范围。
本发明所提供的含烯丙基聚酰亚胺二胺单体:3,3′-二烯丙基-4,4′-二(4-氨基-2-三氟甲基苯氧基)联苯(dbda),其侧链带有烯丙基,分子结构式如下所示:
本发明的含烯丙基二胺单体的制备方法,按照以下步骤进行:
(1)以3,3’-二烯丙基-4,4’-联苯二酚(dabp)和2-氯-5-硝基-三氟甲基苯(cntb)为原料,以无水碳酸钾或碳酸钠为成盐剂,以甲苯或二甲苯为带水剂,二甲基甲酰胺或二甲基乙酰胺作为溶剂,在125~145℃带水5h后,将带水剂蒸出;升温至140~160℃反应10h,混合液出料至冰去离子水中,用水洗涤,得到硝基单体3,3′-二烯丙基-4,4′-二(4-硝基-2-三氟甲基苯氧基)联苯(dnpb)其分子结构式如下所示。其中dabp、cntb和成盐剂三者的摩尔比为1:2.05~2.2:1.05~1.25。
dnpb
上述方法中的3,3’-二烯丙基-4,4’-联苯二酚(dabp),其合成方法参见文献j.poly.sci.partb:poly.phys.,1998,362317,其结构式如下:
(2)将dnpb与fe粉的按照摩尔比为1:10~20加入反应容器,再加入由蒸馏水、乙醇和丙酮三者组成的混合溶剂,在氮气保护下,搅拌加热至回流温度后,将稀盐酸缓慢加入,调节ph为3~5,继续反应5~8h后停止加热。然后加入氢氧化钠以调节ph=8~9,搅拌均匀,将产物趁热过滤,用乙醇水重结晶,60℃真空烘干得到黄色产物dbda。
本发明制备的含烯丙基可交联聚酰亚胺的分子结构式如下所示:
其中ar为
本发明通过亲核缩聚路线,以dbda为主要原料和4,4’-(六氟异丙基)邻苯二甲酸酐(6fda)等进行聚合反应,制备了一系列侧链含烯丙基的聚酰亚胺均聚物和共聚物,从而将可交联的烯丙基引入了聚合物主链结构中,所得的均聚物和共聚物数均分子量mn为83629~92651,重均分子量mw为121227~139758。
制备的含烯丙基可交联聚酰亚胺均聚物及共聚物的聚合反应式为:
其中ar为
本发明的含烯丙基的可交联聚酰亚胺的制备方法,具体方法如下:
以两种二胺单体和4,4’-(六氟异丙基)邻苯二甲酸酐(6fda)为原料,两种二胺单体中一种为dbda,另一种为二胺单体h2n-ar-nh2,以二甲基乙酰胺(dmac)为溶剂,常温搅拌24h形成粘稠聚酰胺酸溶液。再向上述溶液中加入催化剂进行脱水环化反应,然后在60℃油浴中反应8h后,将产物倒入无水乙醇中,出现白色絮状物,过滤后再用蒸馏水和乙醇洗涤,经真空干燥得到含烯丙基的可交联聚酰亚胺共聚物。所述催化剂为体积比为1:2的吡啶和乙酸酐。
所述的二胺单体种h2n-ar-nh2选自4,4`-二氨基二苯醚、2,2`-双(4-氨基苯基)丙烷、4,4`-二氨基二苯砜和3,3`-二氨基二苯甲酮中的一种。
当只采用dbda作为二胺单体进行上述聚合反应时,所得为含烯丙基的可交联聚酰亚胺均聚物。
本发明的含烯丙基可交联聚酰亚胺的用途之一,是用于制备含烯丙基可交联聚酰亚胺膜或含烯丙基可交联聚酰亚胺光交联膜。
其中,制备含烯丙基可交联聚酰亚胺膜的具体方法如下:
按照1g/ml的比例将聚合物溶于n-甲基-2-吡咯烷酮(nmp)中,搅拌至聚合物充分溶解,后混合液经0.22μm的聚四氟乙烯纤维束过滤直接浇注至干净的玻璃板上,60℃停留24h后,每4h升温20℃直至100℃后抽真空,每8h升温20℃直至140℃关闭烘箱,降至室温,取出透明聚酰亚胺聚合物薄膜。
制备含烯丙基可交联聚酰亚胺光交联膜的具体方法如下:
聚合物溶液制备过程与上相同,但要向溶液中再加入0.5~5wt%的光引发剂2,4,6-三甲基苯甲酰基-二苯基氧化膦(tpo),搅拌至均质溶液,制膜过程与上相同。将得到的聚酰亚胺薄膜在紫光灯下照射0.1~1h,即得到光交联聚酰亚胺膜。
本发明具有以下优点:
1、该类聚合物在交联前可溶于普通溶剂中,交联后则发生溶胀或不溶,并且交联过程中无小分子逸出、不易产生空隙,得到的交联产物玻璃化转变温度提高,热稳定性提高。
2、通过调整含烯丙基单体的比例,可制备交联度可控的聚酰亚胺材料,具有较好的热稳定性以及机械性能,拓宽了此类材料的使用范围,同时使其加工方式更加多样化。
附图说明
图1为本发明实施例2制备的二胺单体的红外光谱图;
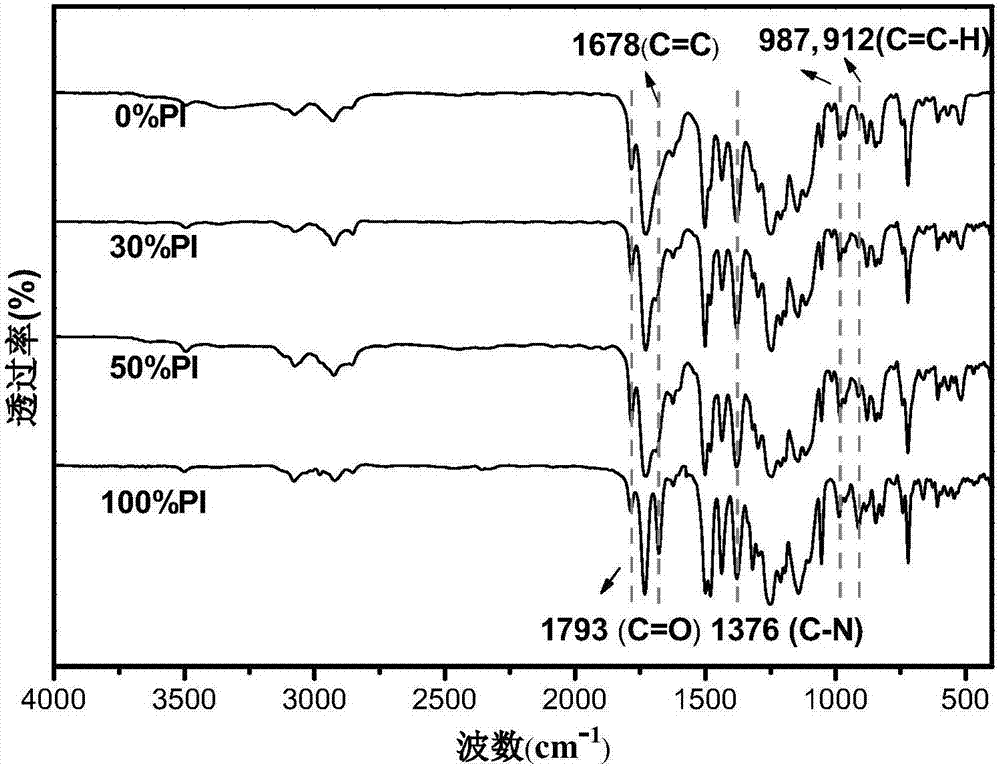
图2为本发明实施例3~实施例6制备的一系列不同dbda含量的含烯丙基可交联聚酰亚胺交联前的红外光谱图;
图3为本发明实施例4~实施例6,实施例8~实施例10所制备的一系列不同dbda含量的含烯丙基可交联聚酰亚胺交联前后的dma曲线。
图4为本发明实施例3~实施例6制备的一系列不同dbda含量的含烯丙基可交联聚酰亚胺交联前后的tga谱图,数据列于表3。
图5为本发明实施例4制备的30%pi和加入1wt%的tpo(光引发剂)的薄膜紫外光照射0.5h后的红外光谱图。
图6为本发明实施例6制备的100%pi的核磁共振氢谱。
具体实施方式
下面通过具体实施方式具体说明本发明的技术方案。
实施例1
(3,3′-二烯丙基-4,4′-二(4-硝基-2-三氟甲基苯氧基)联苯(dnpb)的合成步骤如下:
称取0.01mol烯丙基联苯二酚(dabp),0.0215mol2-氯-5-硝基-三氟甲苯,1.451g碳酸钾(k2co3),加入到带有机械搅拌的100ml三颈烧瓶中,同时再加入30mldmf溶剂和10ml甲苯作为带水剂。在氩气的保护下,当温度达到135℃时保持回流稳定,持续带水3h。放出甲苯,将反应混合溶液温度升高至145℃,反应10h。将反应溶液倒入约500ml的冰去离子水中,得到土黄色沉淀,用蒸馏水常温机械搅拌洗涤数次,抽滤,在真空烘箱中烘干,得到4.8g含烯丙基的二硝基单体(dnpb),产率约为82%。
实施例2
将4.8g(7.4mmol)dnpb加入到250ml三颈瓶中,加入7g(125mmol)还原铁粉,再加入90ml混合溶剂(蒸馏水、乙醇和丙酮各30ml),在氩气保护的条件下,机械搅拌加热至沸腾开始回流。将2ml稀盐酸缓缓滴加到沸腾的三颈瓶中,保持5s/滴。在滴加完hcl之后,反应物继续保持加热回流5h后停止加热。加入0.96g(24mmol)氢氧化钠,搅拌三分钟后,将产物趁热过滤,再用乙醇水重结晶后,保持60℃真空烘干,得到黄色的还原产物含烯丙基二胺单体dbda。所测红外光谱谱图如图1所示。
实施例3
在氮气环境中,将烘干的0.888g(2mmol)6fda加入连有干燥管的100ml三颈瓶中,并用5ml二甲基乙酰胺(dmac)使其完全溶解后,0.4006g(2mmol)oda与13.3mldmac的混合液。滴加完毕后,使混合液常温机械搅拌24h形成粘稠聚酰胺酸。再向混合液中加入2ml吡啶和4ml乙酸酐,在油浴中以60℃的温度,继续反应8h后,出料于无水乙醇中,此时有白色絮状物出现,将其过滤后用蒸馏水和乙醇各洗三遍,在烘箱中真空100℃干燥,得到不含烯丙基的聚酰亚胺(0%pi)。
实施例4
在氮气环境中,将烘干的0.888g(2mmol)6fda加入连有干燥管的100ml三颈瓶中,并用5ml二甲基乙酰胺(dmac)使其完全溶解后,再缓缓滴入0.3505g(0.6mmol)dbda、0.2803g(1.4mmol)oda与10mldmac的混合液。滴加完毕后,使混合液常温机械搅拌24h形成粘性聚酰胺酸。再向混合液中加入2ml吡啶和4ml乙酸酐,在油浴中以60℃的温度,继续反应8h后,出料于无水乙醇中,此时有白色絮状物出现,将其过滤后用蒸馏水和乙醇各洗三遍,在烘箱中真空100℃干燥,得到含30%烯丙基聚酰亚胺聚合物(30%pi)。
实施例5
在氮气环境中,将烘干的0.888g(2mmol)6fda加入连有干燥管的100ml三颈瓶中,并用6ml二甲基乙酰胺(dmac)使其完全溶解后,再缓缓滴入0.5841g(1mmol)dbda、0.2004g(1mmol)oda与10mldmac的混合液。滴加完毕后,使混合液常温机械搅拌24h形成粘性聚酰胺酸。再向混合液中加入2ml吡啶和4ml乙酸酐,在油浴中以60℃的温度,继续反应8h后,出料于无水乙醇中,此时有白色絮状物出现,将其过滤后用蒸馏水和乙醇各洗三遍,在烘箱中真空100℃干燥,得到含50%烯丙基聚酰亚胺聚合物(50%pi)。
实施例6
在氮气环境中,将烘干的1.11g(2.5mmol)6fda加入连有干燥管的100ml三颈瓶中,并用10ml二甲基乙酰胺(dmac)使其完全溶解后,再缓缓滴入1.4605g(2.5mmol)dbda与15mldmac的混合液。滴加完毕后,使混合液常温机械搅拌24h形成粘性聚酰胺酸。再向混合液中加入2.5ml吡啶和5ml乙酸酐,继续反应8h后,出料于无水乙醇中,此时有白色絮状物出现,将其过滤后用蒸馏水和乙醇各洗三遍,在烘箱中真空100℃干燥,得到含100%烯丙基聚酰亚胺聚合物(100%pi),核磁共振谱图如图6所示。
实施例7
将1g聚合物溶于10mln-甲基-2-吡咯烷酮(nmp)中,机械搅拌10小时至聚合物充分溶解,后混合液经0.22μm的聚四氟乙烯纤维束过滤直接浇注至干净的10cm*10cm大小的玻璃板上,60℃停留24h后,每4h升温20℃直至100℃后抽真空,每8h升温20℃直至140℃关闭烘箱,降至室温,取出透明聚酰亚胺聚合物薄膜。
实施例8
将30%pi与n-甲基吡咯烷酮(nmp)按质量体积比为1:10(g/ml)混合,并向其中加入1wt%的光引发剂2,4,6-三甲基苯甲酰基-二苯基氧化膦(tpo),机械搅拌10h至均质溶液,后混合液经0.22μm的聚四氟乙烯纤维束过滤直接浇注至干净的10cm*10cm大小的玻璃板上60℃停留24h后,每4h升温20℃直至100℃后抽真空,每8h升温20℃直至140℃关闭烘箱,降至室温,取出透明聚酰亚胺聚合物薄膜。然后该薄膜在紫光灯下照射0.5h,即得到交联的30%pi,记作c30%pi,dma测试结果如图3所示。
实施例9
将50%pi与nmp按质量体积比为1:10(g/ml)混合均匀,并向其中加入1%tpo,其他步骤同上,得到的交联膜记作c50%pi,dma测试结果如图3所示。
实施例10
将100%pi与nmp按质量体积比为1:10(g/ml)混合均匀后,向其中加入1%tpo,其他步骤同上,得到的交联膜记作c100%pi,dma测试结果如图3所示。
效果检测
测试1(聚合物分子量、比浓粘度测试)
表1聚合物分子量
测试2(机械性能检测)
对光交联膜的机械性能进行了研究,结果如表2所示。从下表中可以看出,与交联之前相比,交联后聚合物膜的拉伸性能有了一定程度的提高,杨氏模量和拉伸强度为均有所提高。
表2聚合物拉伸性能
测试2(tga测试)
表3聚合物在氮气中的热稳定性
如图3所示,发现薄膜交联后的玻璃化转变温度均明显增加,说明烯丙基交联形成三维网状结构,链段运动能力降低。
由图4和表3可知,含有烯丙基的聚合物交联后5%热失重温度都有所增加,说明交联后聚合物分子链形成了三维网状结构从而使得聚合物的热稳定性增加。
由图2和图5的对比可知,经过紫外光照射后膜的双键吸收峰消失,说明烯丙基发生了交联反应。
- 还没有人留言评论。精彩留言会获得点赞!
- 使用乙烯-乙烯基酯系共聚物皂化物的成形材料的制作方法
- 含乙烯-乙烯基酯系共聚物皂化物的成形材料的制作方法
- 乙烯-乙烯基酯系共聚物皂化物粒料和乙烯-乙烯基酯系共聚物皂化物粒料的制造方法
- 一种丙烯酸-1-磺酸丁基-3-乙烯基咪唑硫酸氢盐共聚物及其制备方法和应用
- 一种用于乙烯基甲醚和马来酸酐共聚的聚合釜的制作方法
- 包含活性物和由n-乙烯基内酰胺、乙烯基酯和醇烷氧基化物制成的接枝共聚物的组合物的制作方法
- 一种乙烯基单体反应挤出共聚合的方法
- 包含活性物和由n-乙烯基内酰胺、乙烯基酯和醇烷氧基化物制成的接枝共聚物的组合物的制作方法
- 一种乙烯基单体反应挤出共聚合的方法
- 一种n-取代丙烯酰胺单体、其制备方法、丙烯酰胺共聚物及其制备方法