氮氧化物作为新型组蛋白去乙酰化酶抑制剂的抗肿瘤应用的制作方法
本发明涉及医药技术领域,尤其是涉及氮氧化物作为新型组蛋白去乙酰化酶抑制剂的制备、用途及其衍生物,包含此化合物的药物组合物,涉及所述化合物的制备方法及所述化合物的抗肿瘤应用,涉及所述化合物在疾病,例如抗肿瘤细胞的应用。
背景技术:
hdac抑制剂已经在临床上用于治疗肿瘤疾病,并表现出良好的临床应用前景。其hdac抑制剂对肿瘤细胞主要作用是抑制肿瘤细胞增殖,诱导细胞周期阻滞,诱导肿瘤细胞凋亡和抑制肿瘤新血管的生成等。研究表明hdac抑制剂既可以单独用于治疗肿瘤疾病,也可以与很多其他作用机制的抗肿瘤药物联合使用,都表现出明显的抗肿瘤作用。hdac抑制剂作为一类新型的抗肿瘤药物具有非常重要的研发价值。
目前,hdac抑制剂主要分为五类:异羟肟酸(hydroxamicacid)类,如saha、环氧酮结构的环四肽类如trapoxina、苯甲酰胺(benzamides)类,如ms-275、不含环氧酮结构的环肽类、短链和芳香脂肪酸类,如丁酸钠、苯丁酸钠、杂环化合物类,如depudecin。其中已经上市的hdac抑制剂包vorinostat(saha,zolina)、romidepsin(istodax,fk228,fr901228,depsipeptide、belinostat(beleodaq,pxd-101)、panobinostat(lbh-589,farydak)、西达本胺(chidamide)。根据已经上市的hdac抑制剂的结构特点,其主要包含锌离子鳌合部分(zbg)、连接部分(linker)和表面部分(surfacerecognition,cap)三个部分。zbg部分与活性口袋底部的锌离子进行配位,通常为异羟肟酸、苯甲酞胺、梭酸或亲电酮等。linker主要占据疏水管状通道,以保证其他两部分的正确取向,通常为链状烷烃、肉桂酞基、芳香基或杂环芳香基等。cap与结合口袋入口处的氨基酸残基紧密接触,产生相互作用。以saha为例,其结构如图3所示.
随着分子生物学研究的不断深入,hdac抑制剂的抗肿瘤作用机制逐渐被揭示,但对hdacs亚型是否具有选择性,提高hdac抑制剂的体内稳定性等问题。因此,设计合成氮氧化物作为新型组蛋白去乙酰化酶抑制剂,以已经上市的结构特点为基础,对zbg、linker部分进行修饰,以期发现对hdac亚型具有选择性的hdac抑制剂以及对别的分子靶标的相互作用等。选择抗肿瘤活性的化合物进一步的进行hdac亚型的选择以及对他分子靶标的相互作用。因此,根据羟肟酸类化合物的结构特点进行修饰,主要是探索zbg,linker的变化导致的活性变化。氮氧化合物作为一类新的hdac抑制剂,以期发现对hdac亚型具有良好选择性的化合物,为hdac抑制剂的后续靶向药物开发提供重要理论依据,从而开发出药效更好,特异性更强、选择性更好的hdac抑制剂。
技术实现要素:
本发明的目的是通过氮氧化物作为新型组蛋白去乙酰化酶抑制剂,来寻找对hdac亚型具有良好的选择性的抑制剂。本发明实现该目的的技术方案是提供氮氧化物作为新型组蛋白去乙酰化酶抑制剂及药物组合的制备方法和应用,解决hdac抑制剂对hdac亚型选择性等问题。
为解决上述技术问题,本发明采用下述技术方案:
本发明的氮氧化物作为新型组蛋白去乙酰化酶抑制剂,该化合物的结构通式如下:
其中:n可为1-10个碳;可为不饱和键。
x、z:可同时为c、n;如x=z=n或x=z=c,也可x=c、z=n或x=n、z=c;
r1、r2:c-r11、s、n、o当r1=c时r2可为c、s、n、o其中之一,当r1=c时r2可为c、s、n、o其中之一;
r3、r4:分别为苯环或杂环,其取代基可以为卤素、氨基、羟基、硝基、氰基、1-4个碳原子的烷基、1-4个碳原子的烷氧基、1-4个碳原子的烷氨基、1-4个碳原子的氨烷基、2-4个碳原子的酰基、2-4个碳原子的酰胺基、1-4个碳原子的硫代烷基、1-4个碳原子的全氟烷基、1-4个碳原子的全氟烷或杂环取代基;或分别为氢、羟基;或可同时为二甲胺也可当其中一个为氢或羟基时,另一个为二甲胺(接上二甲胺后再经过氧化得氮氧化合物);
r5:为羟基等;
r6、r7、r8、r9、r10:可以同时有1-4个取代基;当其中之一为卤素、氨基、羟基、硝基、氰基、磺酰基、烷氧基、酰基、2-4个碳原子的酰基、2-4个碳原子的酰胺基、1-4个碳原子的硫代烷基、1-4个碳原子的全氟烷基、1-4个碳原子的氟烷氧基、1-4个碳原子的羧基、1-4个碳原子的烷氧基羧基时;其余分别为卤素、氨基、羟基、硝基、氰基、磺酰基、烷氧基、酰基、2-4个碳原子的酰基、2-4个碳原子的酰胺基、1-4个碳原子的硫代烷基、1-4个碳原子的全氟烷基、1-4个碳原子的氟烷氧基、1-4个碳原子的羧基、1-4个碳原子的烷氧基羧基;其中r6、r7、r8、r9、r10也可以为苯环或杂环;
其中,所述的杂环,是指含一个或多个杂原子(包括氮、氧、硫)的饱和或不饱和杂环,如四氢吡啶、二氢吡诺、二氢吡唑、哌啶、咪唑、吡啶等;
所述的卤素,为氟、氯、溴、碘;
所述的1-4个碳原子的烷基,包括甲基、乙基、正丙基、异丙基、正丁基、异丁基等;
所述的1-4个碳原子的烷氧基,包括甲氧基、乙氧基、正丙氧基、正丁氧基、异丁氧基等;
所述的1-4个碳原子的氨基烷基,包括氨基乙基、1-氨基丙基、1-氨基丙基等;
所述的1-4个碳原子的烷基胺基,包括n-甲胺基、n-乙胺基、n-异丙胺基等;
所述的2-4个碳原子的酰基,包括乙酰基、丙酰基、异丁酰基等;
所述的2-4个碳原子的酰胺基,包括乙酰胺基、丙酰胺基、丁酰胺基、异丁酰胺基等;
所述的1-4个碳原子的硫代烷基,包括甲硫基、乙硫基、丙硫基等;
所述的1-4个碳原子的全氟烷基,包括三氟甲基、五氟乙基等;以氮氧化物作为新型组蛋白去乙酰化酶抑制剂的制备方法包括步骤如下:
步骤1:为酰胺反应。在碱性试剂作用下,使用苯胺与不同链长度的溴化物反应,制备获得2a‐c,其中n=4、6、8;所述的碱性试剂为diea,催化剂为dcc,溶剂为四氢呋喃,反应温度为室温,反应时间约5小时;
步骤2:2a‐c分别与l-脯氨醇、二苯基脯氨醇反应下之别获得3a‐f,其中r=h、ph;采用k2co3使反应处于碱性条件,加入适量nai可让反应产率提高,反应体系采用dmf作为溶剂。反应温度为50-70℃,约反应6个小时;
步骤3:此步为氧化反应,化合物3a‐f在氧化剂的作用下,制备获得目标化合物4a‐f,即式(i)化合物,反应温度为冰浴反应,溶剂为四氢呋喃,氧化剂为mcpba,时间约为3个小时。
其中,实验本采用化合物2b、2c的合成方法来合成2a,但总是不成功,因此本发明改进将4-溴丁酸变为4-溴丁酰氯再与苯胺反应得到2a,反应式如下:2a的化合物为的制备方法如下:
本发明制得的氮氧化物作为新型组蛋白去乙酰化酶抑制剂对人肺癌细胞(a549),人乳腺癌细胞(mcf7)等多种肿瘤细胞生长抑制均较好,可应用于制备预防或治疗肿瘤的药物。
附图说明
图1是hdac抑制剂的筛选示意图;
图2是胰酶抑制剂的筛选示意图;
图3是saha结构示意图。
具体实施方式
下面结合附图和实施例对本发明的技术方案作进一步的详细说明。
氮氧化物作为新型组蛋白去乙酰化酶抑制剂,具有以下结构式(i)、式(ⅱ)化合物:
其中:r选自h或ph;n=4、6、8。
式(i)化合物的制备方法包括如下步骤:
步骤1:为酰胺反应。在碱性试剂作用下,使用苯胺与不同链长度的溴化物反应,制备获得2a‐c,其中n=4、6、8;所述的碱性试剂为diea,催化剂为dcc,溶剂为四氢呋喃,反应温度为室温,反应时间约5小时;
步骤2:2a‐c分别与l-脯氨醇、二苯基脯氨醇反应下之别获得3a‐f,其中r=h、ph;采用k2co3使反应处于碱性条件,加入适量nai可让反应产率提高,反应体系采用dmf作为溶剂。反应温度为50-70℃,约反应6个小时;
式(ⅱ)化合物的制备方法包括如下步骤:
步骤1:为酰胺反应。在碱性试剂作用下,使用苯胺与不同链长度的溴化物反应,制备获得2a‐c,其中n=4、6、8;所述的碱性试剂为diea,催化剂为dcc,溶剂为四氢呋喃,反应温度为室温,反应时间约5小时;
步骤2:2a‐c分别与l-脯氨醇、二苯基脯氨醇反应下之别获得3a‐f,其中r=h、ph;采用k2co3使反应处于碱性条件,加入适量nai可让反应产率提高,反应体系采用dmf作为溶剂。反应温度为50-70℃,约反应6个小时;
步骤3:此步为氧化反应,化合物3a‐f在氧化剂的作用下,制备获得目标化合物4a‐f,即式(i)化合物,反应温度为冰浴反应,溶剂为四氢呋喃,氧化剂为mcpba,时间约为3个小时。
其中,化合物2a反应式和制备方法如下:
实验本采用化合物2b、2c的合成方法来合成2a,但屡次不成功,因此改进将4-溴丁酸变为4-溴丁酰氯再与苯胺反应得到2a,冰浴反应,时间约为4小时,溶剂为四氢呋喃。
本发明化合物抗肿瘤活性的实验方法与结果如下:
(1)组蛋白去乙酰化酶为靶标的氮氧化合物的抗肿瘤试剂对组蛋白去乙酰化酶活性的测定,采用荧光方法检测:
培养a549细胞,从中提取核蛋白,获取含hdac的活性生物样本,进行活性测试,步骤如下:
实验方法:
hdac抑制剂的筛选
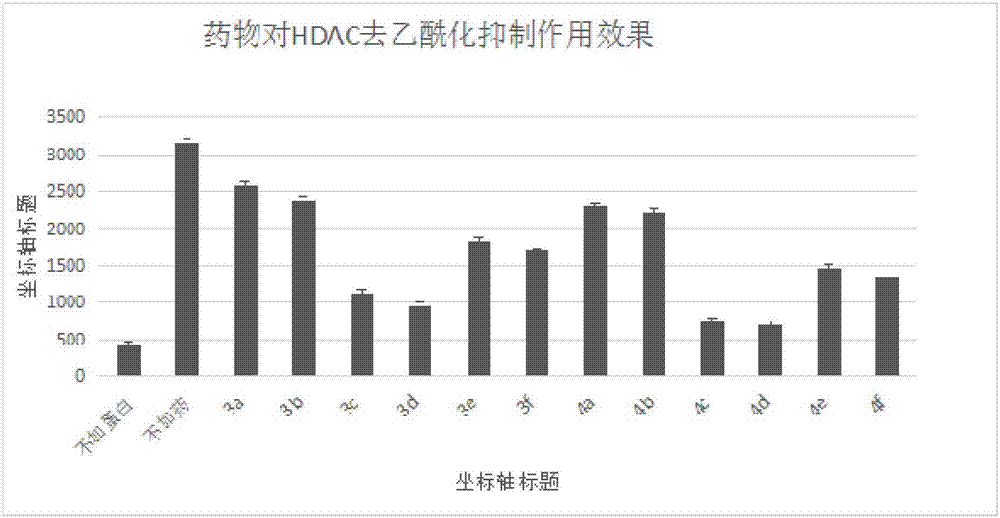
在ep管中加入反应体系需要的溶液(两控制组也要加入相应的dmso),最后加核提取物37℃孵育1h,(第一个样品加入后间隔30s,再加入第二个样品中),反应完成后,30s取一个插冰上,再加入2×胰酶继续孵育1h,反应完成后,30s取一个插冰上,再取120ul加入96孔板中,360nm,460nm处测荧光。药物浓度为100um/ml,部分数据见图1。
从实验结果中可以看出3c、3d、4c、4d在药物浓度为100um的时候对hdac的抑制效果较好,可能存在潜在的hdac抑制剂。由于本实验涉及两步反应,不确定是抑制了hdac还是胰酶,所以下一步针对是否是胰酶的方面的原因,进行了胰酶抑制筛选的实验。
胰酶抑制剂的筛选:
在ep管中加入反应体系需要的溶液(两控制组也要加入相应的dmso),胰酶50ul(不加蛋白组加50ul水),37℃孵育1h(第一个样品加入后间隔30s,再加入第二个样品中),反应完成后,30s取一个插冰上,再取120ul加入96孔板中,在360nm,460nm处使用酶标仪检测荧光。药物浓度为100um/ml。部分数据见图2。
根据如图结果所示,虽然胰酶对该批药物有轻微抑制作用,但是经过图1图2对比可知该批药物对hdac有抑制作用。
(2)氮氧化物作为新型组蛋白去乙酰化酶抑制剂在抗肿瘤细胞的活性测试
采用公认的可用于大规模抗肿瘤药物筛选、细胞毒性试验测定的四甲基氮唑蓝比色法(mtt)评价目标化合物对5种人癌细胞株的抗增殖活性。阳性对照药为临床上广泛使用的抗肿瘤药物阿霉素(adr)及vorinostat(saha)。
细胞株:人肺癌细胞a549,人乳腺癌细胞mcf7,人肝癌细胞hepg2,人结肠癌细胞sw480,脑胶质瘤细胞u87,宫颈癌细胞hela。
细胞增殖抑制率=(阴性对照组od值—药物组od值)*100%/阴性对照组od值。
表1为本发明化合物对肿瘤细胞增殖的抑制率%(100μmol/l)部分实验测试结果如表一:
*:saha的浓度为10μmol/l;其余化合物浓度为100μmol/l。
由实验数据可知所有化合物与阳性saha对照,所有的化合物的抑制剂率都相对偏低,其中3a、3b、4a、4b、3e、3f、4e、4f对a549、mcf7、hepg2、u87、hela肿瘤细胞抑制效率较其他化合物相对较好;3a、3b、3c、3d、3e、3f对a549、mcf7、hepg2、u87、hela肿瘤细胞抑制效率与阳性对照药物saha的抑制率相对偏低,但与saha的抑制率相近。3a、3b、3c、3d、3e、3f分别与4a、4b、4c、4d、4e、4f相比抑制剂率相对偏低,说明氧化后的化合物相对未氧化的化合物抑制剂率较好;4a、4b、4c、4d、4e、4f的抑制率相比,说明linker的长短影响化合物的抑制剂率。故该类化合物具有较大的发展潜力。进一步考察linker部分的长短及结构修饰,有望能得到活性较好的化合物。
综上所述,本发明的优点在于:本发明提供了一种氮氧化合物作为新型hdac抑制剂的抗肿瘤应用及其制备方法,根据活性测试实验数据显示:该类化合物对hdac有潜在抑制剂效果;因此,可进一步在zbg部分提供更多的氮氧、linker进行多个长度的考察等在现有的基础上进行结构修饰,以期发现对hdac亚型具有良好选择性的抑制剂及其他靶点的作用。本发明合成原料相对便宜、工艺简便、纯度较高、成本较低等优点,有望成为对hdac亚型选择性强、高效低毒的新型hdac抑制剂类抗肿瘤药物。
当然,以上只是本发明的具体应用范例,本发明还有其他的实施方式,凡采用等同替换或等效变换形成的技术方案,均落在本发明所要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种含有组氨酸去乙酰化酶抑制剂衍生物的抗肿瘤药物的制造方法与工艺
- 氨甲酰基吡啶酮HIV整合酶抑制剂的方法和中间体与流程
- 氨甲酰基吡啶酮HIV整合酶抑制剂的方法和中间体与流程
- 一类组蛋白去乙酰化酶抑制剂及其制备方法和用途与流程
- 组蛋白去乙酰化酶抑制剂于改变蜂王浆中MRJP3蛋白质的新颖用途的制造方法与工艺
- 用于治疗疾病的硫酸乙酰肝素生物合成抑制剂的制造方法与工艺
- 酚氧类氮氧化物作为去乙酰化酶抑制剂的抗肿瘤应用的制造方法与工艺
- 作为NADPH氧化酶抑制剂的酰氨基噻二唑衍生物的制造方法与工艺
- 利用组蛋白脱乙酰酶抑制剂治疗白血病的制造方法与工艺
- 组蛋白去乙酰化酶HDAC6的抑制剂在制备防治急性肾损伤药物中的用途的制造方法与工艺
- 含有色氨酸基本骨架的邻苯二胺类选择性组蛋白去乙酰化酶抑制剂及其制备方法和应用
- 一种生产贝利司他中间体的工艺方法
- 组蛋白去乙酰化酶抑制剂治疗β亚科疱疹病毒的新用图
- 一种含有苯甘氨酸的肉桂酰胺类组蛋白去乙酰化酶抑制剂及其制备方法和应用
- 2-(1-(4-氯苯甲酰基)-5-甲氧基-2-甲基-1氢-吲哚-3-基)-n-羟基乙酰胺作为组蛋白去 ...的制作方法
- Hif羟化酶抑制剂的药物制剂的制作方法
- 一种组蛋白去乙酰酶抑制剂2-(1-(4-氯苯甲酰基)-5-甲氧基-2-甲基-1氢-吲哚-3-基-n ...的制作方法
- 一种组蛋白去乙酰酶抑制剂(e)-3-(2-(1-(4-氯苯甲酰基)-5-甲氧基-2-甲基-1氢-吲哚 ...的制作方法
- 广谱的去乙酰化酶抑制剂及其应用
- 广谱的去乙酰化酶抑制剂乙酰化赖氨酸及其应用
- 一种制备自噬小体型瘤苗的培养基添加剂及其制备方法与制造工艺
- N-萘烷基取代的含氮杂环衍生物、制备方法及其抗肿瘤用途与制造工艺
- 作为BET蛋白抑制剂的1H‑吡咯并[2,3‑c]吡啶‑7(6H)‑酮和吡唑并[3,4‑c]吡啶‑7(6H)‑酮的制造方法与工艺
- 一种人肿瘤抑制蛋白变体及其应用的制造方法与工艺
- 一种人肿瘤抑制蛋白变体及其应用的制造方法与工艺
- G蛋白信号调节因子6及其抑制剂在治疗脂肪肝和Ⅱ型糖尿病中的功能和应用的制造方法与工艺
- 蛋白酶体抑制剂在制备降低动物脂含量或脂储积能力产品中的应用的制造方法与工艺
- GINS2基因或蛋白的抑制剂在制备抗肿瘤药物中的应用的制造方法与工艺
- ZNF383蛋白在制备抑制p53蛋白活性的产品中的应用的制造方法与工艺
- 蛋白酶体抑制剂MLN9708的合成方法与制造工艺