一种高纯度依帕列净的制备方法与流程
本发明属于药物制备
技术领域:
,具体地说,涉及一种高纯度依帕列净制备方法,使用该方法制备依帕列净,所得产品纯度高,杂质易于控制。
背景技术:
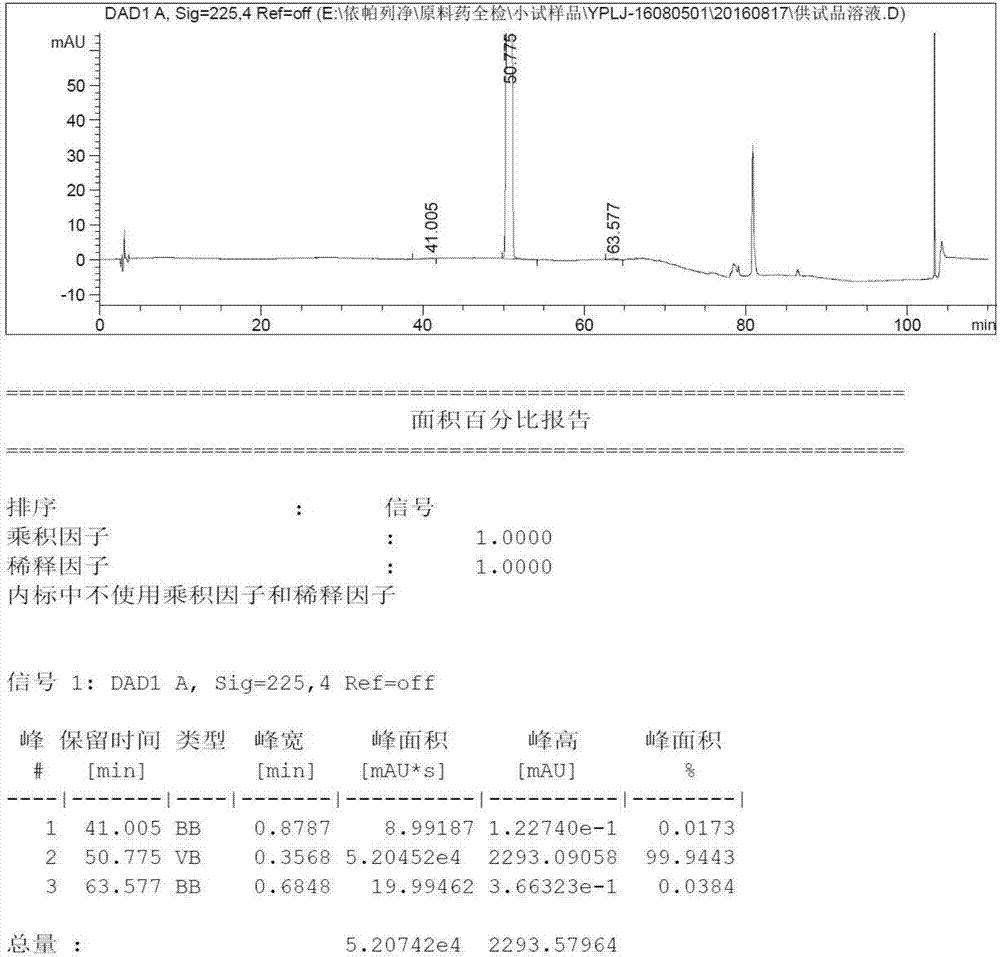
:勃林格殷格翰-礼来糖尿病联盟2014年8月1日联合宣布,糖尿病新药jardiance(empagliflozin,中文名依帕列净)获fda批准,结合饮食及运动用于ⅱ型糖尿病成人患者的治疗,以改善血糖控制。依帕列净化学结构式:中国专利cn102574829a中,描述了一种依帕列净制备方法,该方法是在一定时间内,于-21至-15℃将异丙基氯化镁/氯化锂溶液加至碘代物(化合物1)的四氢呋喃溶液中,经检测反应完全后,加入化合物2;反应完成后加入柠檬酸水溶液,蒸除溶剂后得到化合物3a,再进行缩醛制备(化合物4),三乙基硅烷/三氯化铝还原,反应完成后,在1小时30分钟内于20至30℃加入水(1263l)且在30至53℃于大气压下部分蒸馏混合物,并分离相。将甲苯加至有机相中,且在22至33℃于减压下蒸馏除去溶剂。然后通过在31℃添加晶种使产物结晶,并在冷却至20℃后加入水。在55分钟内冷却反应混合物至5℃,并在3至5℃搅拌12小时。最后离心收集呈无色、结晶固体的产物,用甲苯清洗且在22至58℃干燥。获得产物依帕列净。但在实际重复实验中,本发明人发现该方法操作繁琐,所得产品纯度低(hplc纯度约80%),且用常规纯化方法(重结晶,打浆等),多次纯化仍不能达到合格原料药的标准,且有未知的顽固性杂质不能有效除去。技术实现要素:本发明人对依帕列净的制备方法进行了研究,发现了一种操作简单、适于工业生产的高纯度依帕列净的制备方法,克服了现有技术中存在的不足。本发明的目的是提供一种高纯度依帕列净的制备方法。在本发明的实施方案中,本发明提供了一种高纯度依帕列净的制备方法,包括以下步骤:步骤a.在氮气保护下,将化合物1和2溶于四氢呋喃中,溶解后将体系降至-30℃~-20℃;滴加异丙基氯化镁/氯化锂的四氢呋喃溶液,在-30℃~-20℃反应;反应结束后,加入柠檬酸水溶液,搅拌分液,有机相浓缩完成后,加入有机溶剂,搅拌后,抽滤,得到化合物3;步骤b.将化合物3加入反应釜中,加入甲醇,滴加浓盐酸,搅拌溶解澄清,室温反应;反应完成后,用10重量%的碳酸氢钠水溶液调节ph至中性,乙酸乙酯萃取两次,合并有机相;旋转蒸发脱去有机相溶剂,得黄色油状液体化合物4;步骤c.化合物4溶于乙腈与二氯甲烷的混合溶剂中备用;在干燥的反应釜中加入乙腈、二氯甲烷,加入三氯化铝,三乙基硅烷后,将化合物4的乙腈和二氯甲烷溶液滴加入该反应釜中,搅拌反应,hplc监测反应完成后,加水淬灭反应,搅拌至析出固体,抽滤,收集滤饼,得依帕列净即化合物5粗品;步骤d.粗品精制:将依帕列净粗品加入烷醇类溶剂与烷烃类溶剂的混合溶剂、或者烷醇类溶剂与水的混合溶剂中,加热溶解后,搅拌析晶,抽虑得依帕列净精品,hplc检测,若不合格,重复该操作至合格。在本发明的实施方案中,本发明提供的一种高纯度依帕列净的制备方法,其中,步骤a中所述的有机溶剂为烷醇类溶剂或烷烃类溶剂中的一种,或两者的混合溶剂;所述的烷醇类溶剂为甲醇、乙醇、正丙醇、异丙醇、或正丁醇;所述的烷烃类溶剂为正戊烷、正己烷、或正庚烷,优选地,为正庚烷。在本发明的实施方案中,本发明提供的一种高纯度依帕列净的制备方法,其中,步骤a中柠檬酸水溶液的浓度为1-10重量%,优选地为5-10重量%。在本发明的实施方案中,本发明提供的一种高纯度依帕列净的制备方法,其中,步骤a中,化合物1和化合物2的摩尔比例是1/1.1至1/1.5,优选1/1.3;化合物1与异丙基氯化镁/氯化锂的摩比例是1/1.1至1/1.5,优选1/1.3。在本发明的实施方案中,本发明提供的一种高纯度依帕列净的制备方法,其中,步骤c中,化合物4和三氯化铝的摩尔比例是1/1.1至1/5,优选1/3;化合物4和三乙基硅烷的摩尔比例是1/1.1至1/5,优选1/2.7。在本发明的实施方案中,本发明提供的一种高纯度依帕列净的制备方法,其中,步骤d中所述的烷醇类溶剂为甲醇、乙醇、正丙醇、异丙醇、正丁醇;所述的烷烃类溶剂为正戊烷、正己烷、正庚烷;优选地,所述烷醇类溶剂与烷烃类溶剂的混合溶剂为乙醇和正庚烷的混合溶剂;优选地,所述烷醇类溶剂与水的混合溶剂为甲醇与水的混合溶剂、或乙醇与水的混合溶剂;所述的混合溶剂中烷醇类溶剂与烷烃类溶剂、或者烷醇类溶剂与水的体积比例是1/1至1/2,优选地为1/1至1/1.5。本发明人发现,化合物1与化合物2反应后,产物用柠檬酸水溶液处理后的结构并不是化合物3a,而是化合物3,且化合物3与化合物3a相比产品性状好,易于纯化,因此,本发明的方法制备依帕列净,可得到高纯度产品。另一方面,本发明的方法优化了制备化合物3(步骤a)的投料方式,克服了中国专利cn102574829a需要检测两次的操作繁琐的缺陷;同时,本发明的方法是将化合物1和2混合后,滴加格氏试剂,可以减少投料次数及检测次数,可有效避免多次投料及取样而破坏无水无氧环境,减少杂质的产生。第三发面,本发明的方法优化了步骤c的后处理方式,现有技术cn102574829a中后处理极其繁琐,且所得产品纯度低,不适合工业大量生产;本发明人经过研究后发现,还原反应完成后,加入水淬灭反应,室温搅拌一定时间后即可析出白色固体,抽虑干燥后既得高纯度的依帕列净粗品(>95%),该方式简单易操作,适合工业生产。附图说明图1表示的是按照中国专利cn102574829a实施例5a的方法制备依帕列净成品的hplc谱图,43min杂质超标,且多次精制不能有效除去。图2表示的是采用本发明实施例1和2制备的化合物(3)合成的依帕列净粗品,并且用实施例5的方法精制的依帕列净的hplc谱图,其中无43min杂质,hplc纯度99.94%。具体实施方式下面的实施例是为了详细描述本发明的具体实施方式,而不是为了任何限制本发明的保护范围。实施例1(2s,3r,4s,5r,6r)-2-(4-氯-3-(4-(((s)-四氢呋喃-3-基)氧)苄基)苯基)-3,4,5-三((三甲基硅)氧)-6-(((三甲基硅)氧)甲基)四氢-2h-吡喃-2-醇的合成(3)在氮气保护下,将无水四氢呋喃(2.0l)、化合物1(1.0kg,2.4116mol)和化合物2(1.46kg,3.1350mol)加入到10l玻璃反应釜中,搅拌降温至-30℃~-20℃,向反应釜中滴加iprmgcl/licl(2.4l),控制温度-30℃~-20℃,反应hplc监测完成后,将配置好的5%柠檬酸水溶液滴加到反应釜中,加完后下搅拌30min,静置30min,分液,收集有机相。有机相减压浓缩,浓缩至无冷凝液滴出,收集浓缩物,浓缩后加入2.2l正庚烷室温下打浆5h后抽滤,得类白色粉末状固体,取样检测,hplc纯度98.5%。(安捷伦液相色谱仪,使用十八烷基硅烷键合硅胶为填充剂的色谱柱,磷酸氢二钾溶液/甲醇为流动相梯度洗脱)实施例2(2s,3r,4s,5r,6r)-2-(4-氯-3-(4-(((s)-四氢呋喃-3-基)氧)苄基)苯基)-3,4,5-三((三甲基硅)氧)-6-(((三甲基硅)氧)甲基)四氢-2h-吡喃-2-醇的合成(3)在氮气保护下,将无水四氢呋喃(2.0l)、化合物1(1.0kg,2.4116mol)和化合物2(1.46kg,3.1350mol)加入到10l玻璃反应釜中,搅拌降温至-30℃~-20℃,向反应釜中滴加iprmgcl/licl(2.4l),控制温度-30℃~-20℃,反应hplc监测完成后,将配置好的10%柠檬酸水溶液滴加到反应釜中,加完后下搅拌30min,静置30min,分液,收集有机相。有机相减压浓缩,浓缩至无冷凝液滴出,收集浓缩物,浓缩后加入5l正庚烷与甲醇的混合溶剂(体积比2/1)室温下打浆5h后抽滤,得类白色粉末状固体,取样检测,hplc纯度99.0%。(安捷伦液相色谱仪,使用十八烷基硅烷键合硅胶为填充剂的色谱柱,磷酸氢二钾溶液/甲醇为流动相梯度洗脱)实施例31-c-[4-氯-3-[[4-[[(3s)-四氢-3-呋喃基]氧基]苯基]甲基]苯基]-d-吡喃葡萄糖苷(4)将实施例1或2制备的(3)1.0kg加入反应釜中,加入2.1l甲醇,滴加浓盐酸,搅拌溶清,室温反应4h;反应完成后,用10%碳酸氢钠水溶液调节ph至中性,乙酸乙酯萃取两次,合并有机相;旋转蒸发脱去有机相(40℃±5℃),旋至无冷凝液滴滴下,得黄色油状液体,hplc纯度92.0%.(安捷伦液相色谱仪,以十八烷基硅烷键合硅胶为填充剂的色谱柱,以三氟乙酸水溶液/乙腈为流动相进行梯度洗脱)实施例4(1s)-1,5-脱水-1-c-[4-氯-3-[[4-[[(3s)-四氢-3-呋喃基]氧基]苯基]甲基]苯基]-d-葡萄糖醇(5)将600.0g实施例3制备的化合物(4)溶于1.5l乙腈、1.5l二氯甲烷的混合溶剂中备用。在干燥的反应釜中加入1.5l乙腈、1.5l二氯甲烷,加入550.0g三氯化铝,445.0g三乙基硅烷,将化合物(4)的乙腈和二氯甲烷溶液滴加入上述体系中,搅拌反应,hplc监测反应完成后加入4.0l水,搅拌过夜,抽滤,收集滤饼,得类白色固体(依帕列净粗品),hplc检测,纯度98.6%.(安捷伦液相色谱仪,以十八烷基硅烷键合硅胶为填充剂的色谱柱,以三氟乙酸水溶液/乙腈为流动相进行梯度洗脱)实施例5依帕列净粗品精制反应釜中加入实施例4制备的粗品415.0g,乙醇1l,水1.5l,搅拌均匀后升温至回流,溶解后热滤,滤液室温下搅拌析晶,析出晶体后搅拌2h抽滤,滤饼鼓风干燥,得类白色固体,hplc检测。(安捷伦液相色谱仪,以十八烷基硅烷键合硅胶为填充剂的色谱柱,以三氟乙酸水溶液/乙腈为流动相进行梯度洗脱)实施例6依帕列净粗品精制反应釜中加入实施例4制备的粗品415.0g,乙醇1l,正庚烷1.5l,搅拌均匀后升温至回流,溶解后热滤,滤液室温下搅拌析晶,析出晶体后搅拌2h抽滤,滤饼鼓风干燥,得类白色固体,hplc检测。(安捷伦液相色谱仪,以十八烷基硅烷键合硅胶为填充剂的色谱柱,以三氟乙酸水溶液/乙腈为流动相进行梯度洗脱)实施例7依帕列净粗品精制反应釜中加入实施例4制备的粗品415.0g,甲醇1l,水1l,搅拌均匀后升温至回流,溶解后热滤,滤液室温下搅拌析晶,析出晶体后搅拌2h抽滤,滤饼鼓风干燥,得类白色固体,hplc检测。hplc检测方法安捷伦液相色谱仪,以十八烷基硅烷键合硅胶为填充剂的色谱柱。化合物3供试品浓度为1mg/ml,溶剂为甲醇;使用十八烷基硅烷键合硅胶为填充剂的色谱柱,以5mmol/l的磷酸氢二钾溶液(用磷酸调ph至7.0)为流动相a,以甲醇为流动相b,按下表行梯度洗脱;检测波长为225nm;流速为1.0ml/min,柱温:25℃;进样量20μl。按面积归一化法计算纯度。化合物4供试品浓度为1mg/ml,溶剂为甲醇;以十八烷基硅烷键合硅胶为填充剂的色谱柱;以0.005%三氟乙酸水溶液为a相,以乙腈为b相按下表行梯度洗脱;检测波长为225nm;流速为1.0ml/min;柱温为35℃;进样量10μl。按面积归一化法计算纯度。时间(min)a(%)b(%)07525107525157030305050403565502080555956059560.17525657525依帕列净粗品供试品浓度为1mg/ml,以流动相a-流动相b(75:25)为稀释剂;以十八烷基硅烷键合硅胶为填充剂的色谱柱,以0.005%三氟乙酸水溶液为流动相a,以乙腈为流动相b,按下表行梯度洗脱;检测波长为225nm;流速为1.0ml/min;柱温为25℃;进样量10μl。按面积归一化法计算纯度。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1