一种pH刺激响应性的线形二嵌段聚合前药的制备方法与流程
本发明涉及抗肿瘤药物领域,具体涉及一种ph刺激响应性的线形二嵌段聚合前药的制备方法及其体外活性。
背景技术:
癌症是威胁人类健康的主要疾病之一,其发病率与死亡率仅次于心血管疾病,如何高效的治疗癌症已备受关注。抗肿瘤药物被广泛用于治疗癌症,它能够通过调控dna的复制、转录及翻译有效地抑制肿瘤的增殖并促进癌细胞的凋亡,常用的有甲氨喋呤、吡喃阿霉素、盐酸阿霉素以及从天然产物中提取的喜树碱、紫杉醇等,但是这些化学药物在杀死癌细胞的同时损伤了正常细胞和组织,出现呕吐、高热、脱发、免疫力下降等症状,引起这一系列问题的主要原因是抗肿瘤药物的水溶性差,选择性差从而不能在病变部位有效发挥药效,导致它们的应用在临床上受到限制。
采用物理包埋或者化学键合的方式将抗肿瘤药物与纳米载体结合形成胶束既可以大幅度降低药物的毒副作用,又可以实现药物在病变部位的控制释放。研究表明通过靶向试剂对药物载体的修饰,可以提高药物在肿瘤部位的分布使药物充分发挥作用。cn201610987991.6公开了一种ph-氧化还原双响应性的靶向纳米给药载体(肿瘤靶向配体-聚乙二醇-二硫键-聚酰胺-胺-组氨酸),其中组氨酸的修饰增加了载体的酸敏感性,使聚酰胺-胺的质子海绵效应增强,促进药物溶酶体中快速释放。cn201611196325.7公开了一种ph响应胰岛素缓释的三嵌段聚合物(聚乙二醇单甲醚-聚己内酯-聚碱聚甲基丙烯酸-n,n-二乙氨基乙酯),该纳米颗粒能够根据不同ph值进行药物的精准投递,同时能够达到缓释的效果。
由于到达病变部位的药物浓度仍低于治疗所需剂量,因此,构建药载量高、刺激响应性的靶向性药物载体具有重要的意义。
技术实现要素:
本发明的目的之一是一种ph刺激响应性线形聚合前药的制备方法,该方法操作简单,产率高。目的之二是使用所述制备方法制备的聚合物前药具有药载量高、稳定性好、毒副作用小、ph刺激响应性、叶酸靶向性等优点。为实现上述目的,采用以下技术方案:
1)一种ph刺激响应性的线形二嵌段聚合前药的制备方法,包含以下步骤:
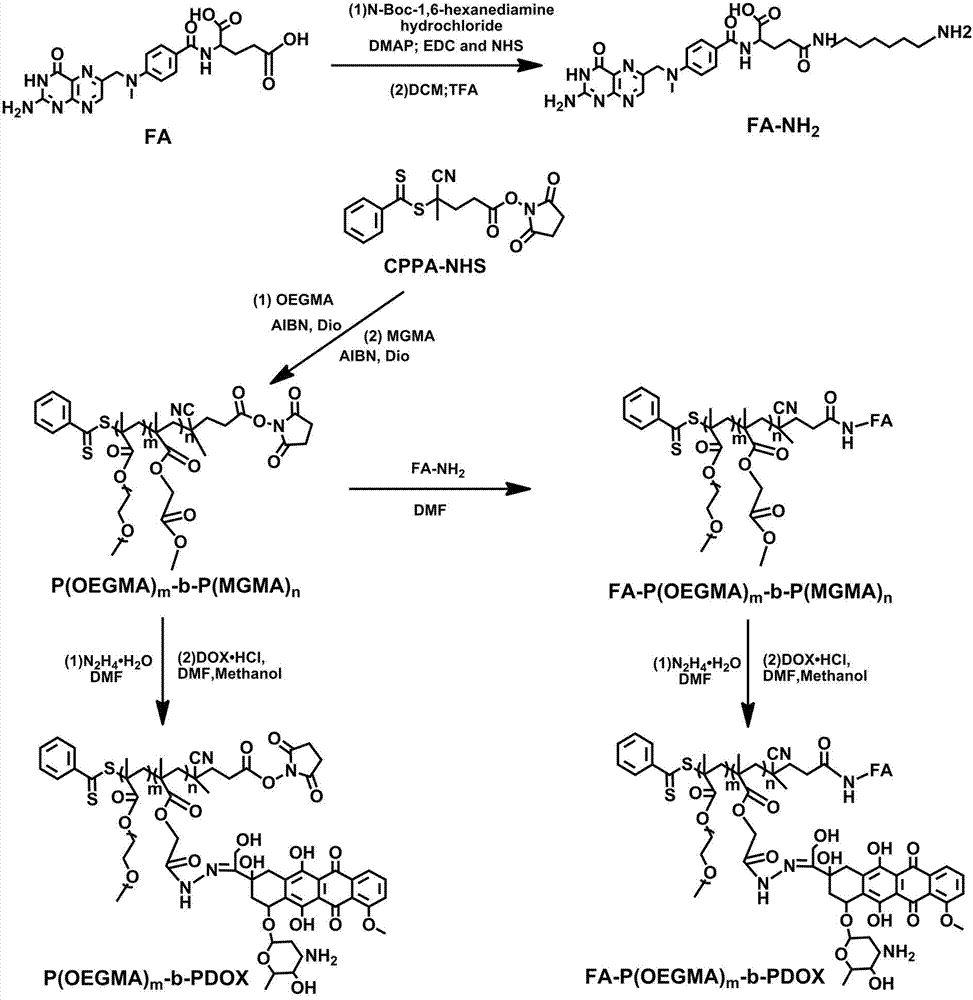
(1)亲水单体p(oegma)m的制备,其反应式如下所示,包含以下步骤:在氩气(ar,2-10pa)条件下,以cppa-nhs为raft反应的大分子引发剂,与乙二醇甲基丙烯酸酯(oegma)一并溶于1,4-二氧六环(dio)中,加入偶氮异丁二腈(aibn)后,冷冻-解冻循环三次,在70℃下避光反应24h,用液氮冷冻以终止反应,解冻,加3~5ml甲醇稀释,用截留分子量(mwco)为3500da的透析袋在甲醇中透析24h,浓缩后得到亲水单体p(oegma)m,其中m表示oegma的聚合度聚合度(dp),它的范围为5~120;
(2)两亲性单体p(oegma)m-b-p(mgma)n的制备,包含以下步骤:
a)mgma的制备,其反应式如下所示,包含以下步骤:在冰浴(温度≤0℃)和氩气(ar,2-10pa)条件下,羟基乙酸甲酯(mg)溶于含三乙胺(tea)的二氯甲烷(dcm)溶液中并搅拌0.5h,然后将甲基丙烯酰氯(ma)溶于二氯甲烷(dcm)所形成的混合液缓慢加入,0.5h后置于室温下避光反应12h,过滤除杂,用旋转蒸发仪浓缩溶剂,进一步以乙酸乙酯、正己烷为洗脱剂过硅胶柱除去二取代产物,用旋转蒸发仪浓缩所收集溶液即得纯的甘氨酸乙酯甲基丙烯酰胺(mgma);
b)p(oegma)m-b-p(mgma)n的制备,其反应式如下所示,包含以下步骤:在氩气(ar,2-10pa)条件下,将步骤(1)制备的p(oegma)m作为raft反应的大分子引发剂,与步骤(2)a)制备的mgma一起溶于dio中,加入aibn后,冷冻-解冻循环三次,在70℃下避光反应24h,用液氮冷冻以终止反应,解冻,加3~5ml甲醇稀释,用mwco为3500da的透析袋在甲醇中透析24h,浓缩后得到两亲性二嵌段聚合物p(oegma)m-b-p(mgma)n,其中n表示mgma的聚合度(dp),n的范围为10~120;
(3)聚合前药p(oegma)m-b-pdox的制备,其反应式如下所示,包含以下步骤:常温(25℃)通氩(ar,2-10pa)条件下,将步骤(2)制备的二嵌段聚合单体p(oegma)m-b-p(mgma)n与单水合肼(n2h4·h2o)溶于n,n-二甲基甲酰胺(dmf)与甲醇形成的混合液中,避光反应12h,用mwco为3500da的透析袋在水中透析24h后冷冻干燥48h得到含酰肼的两亲物p(oegma)m-b-hydrazide;将该物质与盐酸阿霉素(dox.hcl,其结构如下所示),在氩气(ar,2-10pa)条件下溶于含等体积甲醇与dmf的混合液中,加入1滴三氟乙酸(tfa)后避光室温(25℃)反应48h,用mwco为3500da的透析袋在甲醇中透析,用旋转蒸发仪浓缩后即得到聚合前药p(oegma)m-b-pdox(简称为om@dox);
(4)ph刺激响应性聚合前药om@dox纳米胶束的制备,包含以下步骤:取5mg步骤(3)制备的聚合前药om@dox,溶于含tea的dmf混合液中,搅拌0.5h后,缓慢滴加至二次水中并搅拌0.5h,用mwco为3500da的透析袋在水中透析24h除去有机溶剂,即得到纳米胶束水溶液。
进一步的,步骤(1)中aibn、cppa-nhs和oegma的摩尔浓度比为1:(2.4~6.7):(8~24)。
进一步的,所述步骤(2)a)中tea与dcm的体积比范围为1:(2~4.5);所述步骤(2)a)中mg和ma的摩尔浓度范围比为1:(1.7~2.3);所述步骤(2)a)中乙酸乙酯和正己烷的体积比为1:(3.62~4.8);所述步骤(2)b)中p(oegma)m和mgma摩尔浓度比为1:(10~80)。
进一步的,所述步骤(3)中p(oegma)m-b-p(mgma)n、p(oegma)m-b-hydrazide和dox·hcl的摩尔浓度比为1:(1.2~3.6):(1.4~5.1);所述步骤(3)中n2h4·h2o、甲醇、dmf和tfa的体积比为1:(6~10):(2~7):(0.1~0.4)。
进一步的,步骤(4)的聚合前药中阿霉素的浓度范围为0.01mg·l-1~0.05mg·l-1;所述步骤(4)中dmf和水的体积比范围为1:(6~500);所述步骤(4)中透析后所得的纳米胶束粒径范围为10~1000nm。
2)一种ph刺激响应性的线形二嵌段聚合前药的制备方法,该聚合前药还具有靶向性,包含以下步骤:
(i)fa-nh2的制备,其反应式如下所示,包含以下步骤:在冰浴(温度≤0℃)和氩气(ar,2-10pa)条件下,将叶酸(fa)溶于无水的dmf中,加入催化剂1-(3-二甲氨基丙基)-3-乙基碳二亚胺(edc)和n-羟基硫代琥珀酰亚胺(nhs),搅拌0.5h后,加入n-叔丁氧羰基-1,6-己二胺(nh2-boc)溶于含tea的dmf的混合液,在室温下(25℃)反应24h,缓慢滴加至不断搅拌的二次水中,过滤,真空干燥后得到氨基被叔丁氧羰基保护的fa-nh-boc,加入dcm和tfa溶剂并搅拌0.5h,浓缩,加入dcm及tea,避光室温(25℃)搅拌24h,转速设为5000r/min离心3min,用dcm洗两次,真空干燥后得到fa-nh2;
(ii)fa-p(oegma)m-b-p(mgma)n的制备,其反应式如下所示,包含以下步骤:在室温(25℃)通氩(ar,2-10pa)条件下将步骤(2)制备的二嵌段聚合物p(oegma)m-b-p(mgma)n与步骤(i)制备的fa-nh2溶于含tea的无水二甲亚砜(dmso)溶液中,避光反应48h后,缓慢滴加至乙醚中沉淀,真空干燥得到接叶酸的两亲性聚合物fa-p(oegma)m-b-p(mgma)n;
(iii)聚合前药fa-p(oegma)m-b-pdox的制备,其反应式如下所示,包含以下步骤:常温(25℃)通氩(ar,2-10pa)条件下,将步骤(ii)制备的二嵌段共聚物fa-p(oegma)m-b-p(mgma)n与单水合肼(n2h4·h2o)溶于n,n-二甲基甲酰胺(dmf)与甲醇形成的混合液中,避光反应12h,用mwco为3500da的透析袋在水中透析24h后冷冻干燥48h得到含酰肼的两亲物fa-p(oegma)m-b-hydrazide。将该物质与dox·hcl在氩气(ar,2-10pa)条件下溶于含等体积甲醇与dmf的混合液中,加入1滴三氟乙酸(tfa)后避光室温(25℃)反应48h,用mwco为3500da的透析袋在甲醇中透析,用旋转蒸发仪浓缩后即得到聚合前药fa-p(oegma)m-b-pdox,记为fa-om@dox;
(iv)一种ph刺激响应性聚合前药fa-om@dox纳米胶束的制备,包含以下步骤:取5mg步骤(iv)制备的聚合前药fa-om@dox,溶于含tea的dmf混合液中,搅拌0.5h后,缓慢滴加至二次水中并搅拌0.5h,用mwco为3500da的透析袋在水中透析24h除去有机溶剂,即得到纳米胶束水溶液。
进一步的,所述步骤(i)中fa、edc、nhs、nh2-boc和tea的摩尔浓度比范围为1:(1.01~1.2):(1.04~1.3):(1.8~2.4)(1.4~2.2);所述步骤(i)中dcm、tfa和tea的体积比范围为1:(0.92~1.08):(1.8~2.4);
进一步的,所述步骤(ii)中p(oegma)m-b-p(mgma)n、fa-nh2和tea的摩尔浓度比范围为1:(1.01~1.2):(1.01~1.2);
进一步的,所述步骤(iii)中fa-p(oegma)m-b-p(mgma)n、fa-p(oegma)m-b-hydrazide和dox·hcl的摩尔浓度比范围为1:(1.2~3.6):(1.4~5.1);所述步骤(iii)中n2h4·h2o、甲醇、dmf和tfa的体积比范围为1:(6~10):(2~7):(0.1~0.4)。
进一步的,步骤(iv)的聚合前药中阿霉素的浓度范围为0.01mg·l-1~0.05mg·l-1;所述步骤(iv)中dmf和水的体积比范围为1:(6~500);所述步骤(iv)中透析后所得的纳米胶束粒径范围为10~1000nm。
主要优点:
针对聚合前药递送体系现存的问题,本项目提出一类ph刺激响应性的线形二嵌段聚合前药递送体系,该体系有效的提高了药物上载量及药物释放量,降低了对正常细胞的毒副作用,最重要的是,该体系可以实现药物的靶向递送并选择性释放,从而实现癌症的精准治疗。
附图说明
为了更加清楚的展现本发明的目的及其技术方案,本发明提供如下附图。
图1为本发明实施例1中的聚合前药om@dox和实施例2中的聚合前药fa-om@dox的制备流程示意图。
图2为本发明实施例1中的p(oegma)m与p(oegma)m-b-p(mgma)n的核磁示意图。
图3为本发明实施例2中的fa-p(oegma)m-b-p(mgma)n与fa-p(oegma)m-b-pdox的核磁示意图。
图4为本发明实施例1中的聚合前药om@dox纳米胶束的dls和tem示意图。
图5为本发明实施例1中的聚合前药om@dox纳米胶束的释放示意图。
图6为本发明实施例1中的聚合前药om@dox纳米胶束和实施例2中的聚合前药fa-om@dox纳米胶束的毒性示意图。
具体实施方式
下面将结合附图,对本发明的实施例进行详细的描述。
实施例1制备ph刺激响应性的聚合前药
1.制备p(oegma)m:在氩气(ar,2-10pa)条件下,以cppa-nhs为raft反应的大分子引发剂,与乙二醇甲基丙烯酸酯(oegma)一并溶于1,4-二氧六环(dio)中,加入偶氮异丁二腈(aibn)后,冷冻-解冻循环三次,在70℃下避光反应24h,用液氮冷冻以终止反应,解冻,加3~5ml甲醇稀释,用截留分子量(mwco)为3500da的透析袋在甲醇中透析24h,浓缩后以氘代-二甲亚砜为溶剂(dmso-d6)测氢谱结果如图2(a)所示,1(3.24ppm)和2(3.60ppm)分别为oegma上甲氧基和亚甲基氢的信号峰,表明亲水单体p(oegma)m成功合成;
2.制备p(oegma)m-b-p(mgma)n:
a)mgma的制备:在冰浴(温度≤0℃)和氩气(ar,2-10pa)条件下,羟基乙酸甲酯(mg)溶于含三乙胺(tea)的二氯甲烷(dcm)溶液中并搅拌0.5h,然后将甲基丙烯酰氯(ma)溶于二氯甲烷(dcm)所形成的混合液缓慢加入,0.5h后置于室温下避光反应12h,过滤后浓缩即得纯的mgma;
b)p(oegma)m-b-p(mgma)n的制备:在氩气(ar,2-10pa)条件下,p(oegma)m与mgma一并溶于dio中,加入aibn后,冷冻-解冻循环三次,在70℃下避光反应24h,用液氮冷冻以终止反应,解冻,透析24h,浓缩纯化;以氘代氯仿(cdcl3)为溶剂所测的氢谱结果如图2(b)所示,4(3.73ppm)与5(4.55ppm)分别为mgma上甲氧基和亚甲基上氢的信号峰,表明两亲性单体p(oegma)m-b-p(mgma)n成功合成;
3.制备p(oegma)m-b-pdox:常温(25℃)通氩(ar,2-10pa)条件下,p(oegma)m-b-p(mgma)n与单水n2h4·h2o溶于dmf与甲醇形成的混合液中,避光反应12h,透析24h后冷冻干燥48h得到含酰肼的两亲物;将该物质与dox·hcl在氩气(ar,2-10pa)条件下溶于含等体积甲醇与dmf的混合液中,加入1滴三氟乙酸(tfa)后避光室温(25℃)反应48h,透析后浓缩后即得到聚合前药p(oegma)m-b-pdox(om@dox);
4.制备聚合前药om@dox纳米胶束:5mg聚合前药om@dox溶于含tea的dmf混合液中,搅拌0.5h后,缓慢滴加至二次水中并搅拌0.5h,在水中透析24h除去有机溶剂,即得到纳米胶束水溶液;如图4所示,om@dox纳米胶束为球状,多分散指数(pdi)比较小,粒径介于1~100之间,表明所制胶束尺寸均匀易进入细胞;体外释放如图5所示,96h时om@dox纳米胶束在ph=5.0的媒介中释放百分比为90.0%,而在ph=7.4的媒介中为11.4%,表明该体系在酸性条件下趋于完全释放,具有ph刺激响应性;体外毒性如图6所示,om@dox胶束作用72h后,图6a)中宫颈癌细胞(hela)存活率为16.2%,从图6(b)中可以看出对正常细胞的(l929)毒副作用较小。
实施例2制备ph刺激响应性的聚合前药,该聚合前药还具有靶向性
(i)fa-nh2的制备:在冰浴(温度≤0℃)和氩气(ar,2-10pa)条件下,将叶酸(fa)溶于无水的dmf中,加入催化剂edc和nhs,搅拌0.5h后,加入nh2-boc溶于含tea的dmf形成的混合液,在室温下(25℃)反应24h,缓慢滴加至不断搅拌的二次水中,过滤,真空干燥后得到fa-nh-boc。加入dcm和tfa溶剂并搅拌0.5h,浓缩,加入dcm及tea,避光室温(25℃)搅拌24h,转速设为5000r/min离心3min,用dcm洗两次,真空干燥后得到fa-nh2;
(ii)制备fa-p(oegma)m-b-p(mgma)n:在室温(25℃)通氩(ar,2-10pa)条件下p(oegma)m-b-p(mgma)n与fa-nh2溶于含tea的dmso溶液中,避光反应48h后,在乙醚中沉淀,真空干燥后以氘代-二甲亚砜(dmso-d6)为溶剂所测的氢谱结果如图3(a)所示,6.63~8.60ppm叶酸特征峰的出现表明聚合物fa-p(oegma)m-b-p(mgma)n成功合成;
(iii)制备fa-p(oegma)m-b-pdox:常温(25℃)通氩(ar,2-10pa)条件下,二嵌段共聚物fa-p(oegma)m-b-p(mgma)n,与n2h4·h2o溶于dmf与甲醇形成的混合液中,避光反应12h,透析24h后冷冻干燥48h得到含酰肼的两亲物;将该物质与dox·hcl在氩气(ar,2-10pa)条件下溶于含等体积甲醇与dmf的混合液中,加入1滴tfa,避光室温(25℃)反应48h,透析浓缩后,以氘代二甲基亚砜(dmso-d6)为溶剂所测的氢谱结果如图3(b)所示,7.98~7.78ppm阿霉素特征峰的出现表明聚合前药fa-p(oegma)m-b-pdox(fa-om@dox)成功合成,通过核磁结果计算药载量高达61.5%;
(iv)制备聚合前药fa-om@dox纳米胶束:5mg聚合前药fa-om@dox溶于含tea的dmf混合液中,搅拌0.5h后,缓慢滴加至二次水中并搅拌0.5h,在水中透析24h除去有机溶剂,即得到纳米胶束水溶液;通过比较体外毒性图6(c)与图6(d),显然fa-om@dox胶束作用时间相同时,口腔表皮样癌细胞(kb)的存活率低于(~4%)肺癌细胞(a549),表明fa-om@dox胶束具有靶向性。
最后说明的是,以上优选实施例仅用以说明本发明的技术方案而非限制,尽管通过上述优选实施例已经对本发明进行了详细的描述,但本领域技术人员应当理解,可以在形式上和细节上对其做出各种各样的改变,而不偏离本发明权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!