一种制备偶氮金属络合物的方法与流程
本发明属于化学领域,具体为一种制备偶氮金属络合物的方法。
背景技术:
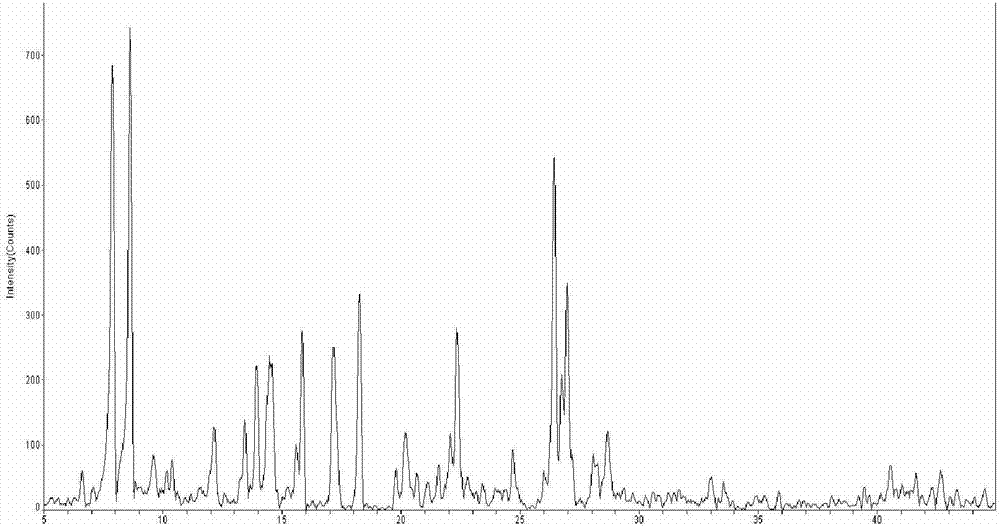
:现有技术中,难溶于水的偶氮金属络合物通常的制备方法是将偶氮中间体、金属赋予剂溶解或分散在有机溶剂中,升温达到溶剂回流状态下反应到预定时间后结束反应,过滤,固体物质再以酸或碱或盐在一定ph条件下进行离子交换,调节偶氮金属络合物外围的反离子,即抗衡离子。专利cn1244641c公布的偶氮铬、cn100419581c、cn1607468a、cn1938386a、cn1759102a、cn1004402504c公布的偶氮铁,在其络合反应过程中,均使用了大量的乙二醇、乙二醇单乙醚、丁醇等有机溶剂,带来一些问题。由于偶氮金属络合物在有机溶剂中具有一定的溶解度,导致络合反应结束后晶体的原生粒径通常都在10微米以上,而作为电荷调节剂使用的粒径通常是在10微米以下,这就增加了粉碎的成本;络合反应过程中使用到的有机溶剂,操作者长期接触后会身体健康受到影响;溶剂的回收也增加了大量的能耗。偶氮金属络合物作为电荷调节剂使用时,络合物外围的反离子是氢离子的时候,其电荷调节性能是理想而且优秀的。通常的方法是络合反应结束后进行固液分离,所得偶氮金属络合物分散在盐酸或硫酸与水组成的酸性反离子交换处理液中,一定温度、时间下高速搅拌分散处理,离子交换反应结束后进行固液分离、洗涤,直至洗涤液的电导率达到要求。探索难溶性偶氮染料的金属络合物的以水代替有机溶剂的合成方法,目前该类合成取得了一定进展。专利cn200810197365.2公开了一种在水系媒体中,偶氮化合物与水杨酸铬钠盐进行配位反应制备偶氮铬络合物的方法。该法是以水-强极性溶剂为混合反应介质,即在水体系中添加季铵盐和冠醚类阳离子表面活性物质,和dmf,dmso等强极性有机溶剂,以及无机强碱来促使铬配位反应进行的。该法虽然对强极性溶媒的量未做权利限制,但强极性溶剂在水中发挥作用,至少用量达到5%以上(以偶氮铬产物重量计,下同),专利说明书上显示季铵盐用量超过6%,dmf、dmso用量超过20%。由于用到的有机溶剂的沸点较高且具有强吸湿性,必须且只能通过水洗涤除去,才能保证产品的电性能。因此洗涤回收废水较多,强极性溶媒进入废水,造成废水中有机物增加。另一方面,该法是在强碱体系下制备偶氮铬电荷控制剂,需要用盐酸中和后酸化置换较大的阳离子,为了更好的置换,加入10%乙醇(见专利描述书),盐酸置换工艺后,进行洗涤,延长了工艺流程,产生较多的含盐废水。山西大学董金龙2006年硕士论文《偶氮金属(铜、锌)络合染料的合成、结构及应用》,采用了乙醇与水体积比为1的混合体系作为反应介质,氯化锌作为金属锌赋予剂,制备得到偶氮锌金属络合物2[zn(c16h13n4o8s)·2h2o·oc2h5],锌络合物收率为78%;但是其配体偶氮化合物不具备邻,邻-二羟基结构,且反应介质使用的是混合有机溶剂。山西大学曹巧风2007年硕士论文《偶氮金属络合染料的合成及表征》,采用与董金龙同样的方法制备了偶氮锌配合[zn(c18h15n2o)(h2o)2]卤原子2,锌络合物收率为75%。该配体偶氮化合物同样不具备邻,邻-二羟基结构,同样采用水和有机溶剂的混合体系作为反应介质。综上所述,制备水难溶性偶氮金属(锌、铬、铁、铜等)络络合物通常使用一定量的溶纤剂类、醇类、dmf、dmso等高沸点极性溶剂,或者这些有机溶剂与水组成的混合溶剂,产生了大量的有机溶剂及废水。随着环境保护要求越来越高,对劳动者保护意识越来越强,难溶于水偶氮金属络合物络合制备工艺还有研究的余地,应朝着清洁生产方向发展。技术实现要素:本发明针对上述存在的问题提供一种易于操作,步骤简单、环保的制备偶氮金属络合物的方法。为达到上述目的,本发明的技术方案如下:本发明的采用水热法制备偶氮金属络合物,用下述通式i的邻,邻’-二羟基偶氮化合物与金属赋予剂经络合反应合成具有下述通式ⅱ结构的1:2型偶氮金属络合物其中,m表示二价或三价金属原子,n为1或2;所述通式ⅰ、通式ⅱ中:ar1表示ar2表示所述的中,r1为h、卤原子、硝基、含1-18个碳原子的直链或支链烷基、含2-18个碳原子的直链或支链烷基、含取代基的磺胺基、甲磺酰基、羟基、含1-18个碳原子的烷氧基、乙酰氨基、苯甲酰氨基或含取代基的芳基;所述的中,r2为萘基或取代萘基;所述的取代萘基上取代基为卤原子、c1-c5烷基或苯基酰胺基;所述的中,r3为h、c1-c18烷基;r4为芳基或取代芳基;所述的取代芳基上的取代基为卤原子、1-5个碳原子的烷氧基、具有1-3个碳原子的羟烷基、2-5个碳原子的烷氧基羰基、1-3个碳原子的烷巯基、1-4个碳原子的烷基氨基、2-8个碳原子的二烷基氨基、氰基、羟基、硝基、氨基、乙酰氨基或磺胺基。进一步,所述的络合反应的反应介质为蒸馏水或去离子水;通式ⅰ的化合物与金属赋予剂中金属离子的投料摩尔比为2-8:1;反应温度为80-300℃,压力为0.05mpa-20.0mpa,反应时间0.5-10小时,通式ⅰ的化合物与金属赋予剂质量之和与反应介质的质量比为1:1-9。进一步,所述的金属赋予剂为含cr3+、fe3+、al3+、zn2+、fe2+、cu2+、co2+、ni2+或mn2+的无机酸盐、有机酸盐或金属有机络合物。进一步,所述的无机酸盐为盐酸盐、硫酸盐、硝酸盐或磷酸盐;所述的有机酸盐为甲酸盐、乙酸盐、丙酸盐、水杨酸盐、乳酸盐、水杨酸金属络合物或氨基酸金属络合物。进一步,所述的m表示cr、三价fe、al、zn、二价fe、cu、co、ni、mn中的一种。进一步,所述的通式ⅱ的络合物直接与na+、k+、nh4+、c2~c16烷基胺离子结合,置换出原来的h+。进一步,所述的通式ⅱ的偶氮金属络合物结构式中,偶氮配体与金属原子的摩尔比为2:1。本发明的通式ⅰ化合物具有邻,邻’-二羟基结构,可以与金属离子形成络合物,例如与zn2+、cu2+、co2+、ni2+、fe2+、mn2+反应形成相应的偶氮二价金属络合物,与cr3+、fe3+、al3+反应形成相应的偶氮三价金属络合物。本发明的偶氮金属络合物与na+、k+、nh4+、c2~c16烷基胺离子结合后,得到以下化合物之一:其中,m为cr、三价fe、al、zn、cu、co、ni、二价fe或mn;n为1或2;a+为na+、k+、nh4+或c2~c16烷基胺离子;其中,m为cr、fe、al、zn、cu、co、ni、fe或mn;n为1或2;a+为na+、k+、nh4+或c2~c16烷基胺离子;其中,m为cr、三价fe、al、zn、cu、co、ni、二价fe或mn;n为1或2;a+为na+、k+、nh4+或c2~c16烷基胺离子;其中,m为cr、三价fe、al、zn、cu、co、ni、二价fe或mn;n为1或2;a+为na+、k+、nh4+或c2~c16烷基胺离子;其中,m为cr、三价fe、al、zn、cu、co、ni、二价fe或mn;n为1或2;a+为na+、k+、nh4+或c2~c16烷基胺离子;其中,m为cr、三价fe、al、zn、cu、co、ni、二价fe或mn;n为1或2;a+为na+、k+、nh4+或c2~c16烷基胺离子;其中,m为cr、三价fe、al、zn、cu、co、ni、二价fe或mn;n为1或2;a+为na+、k+、nh4+或c2~c16烷基胺离子;其中,m为cr、三价fe、al、zn、cu、co、ni、二价fe或mn;n为1或2;a+为na+、k+、nh4+或c2~c16烷基胺离子;其中,m为cr、三价fe、al、zn、al、cu、co、ni、二价fe或mn;n为1或2;a+为na+、k+、nh4+或c2~c16烷基胺离子。本发明还提供一种色调剂,应用了所述的制备方法得到的偶氮金属络合物,所述的偶氮金属络合物的含量为色调剂总重量的0.1-2%。所述的色调剂,其制备方法包括以下步骤:由金属络合物、着色剂、粘结树脂、疏水纳米二氧化硅制备而成,现将偶氮金属络合物、着色剂、粘结树脂同时熔融混炼,冷却至常温以下进行粉碎、筛分,收集8~10微米颗粒,得到混合物:按混合物总重量的0.4-1.5%混入疏水纳米二氧化硅,充分混合即得。所述的制备方法得到的偶氮金属络合物还可以用于聚合法制备色调剂,聚合法制备色调剂的过程中加入本发明制备方法得到的偶氮金属络合物电荷控制剂的方法有两种,即预先往合成色调剂粒子过程中添加,或者在制备完成的色调剂粒子表面添加。本发明的制备偶氮金属络合物的方法所制备的偶氮金属络合物应用于在色调剂中。本发明的有益效果为:本发明经过大量的试验研究发现,以水热法制备偶氮金属络合物,其络合反应介质采用蒸馏水或去离子水,未使用任何有机溶剂,解决了水不溶性或难溶性偶氮配体与金属的络合问题、有机溶剂废水污染、回收问题。当络合物外围反离子为氢离子时,后期无需使用酸或碱来调节络合物外围反离子即可作为电荷调节剂或染料使用;其工艺简单环保,所得产品作为电荷电荷调节剂使用时具有实用而优秀的电荷调节性能。本发明的偶氮金属络合物无需进行额外的反离子调节,在色调剂中作为电荷调节剂具有实用的起电速率和带电量,并且具有良好的热稳定性及树脂相容性,另外,本发明的偶氮二价金属络合物其原生粒径较小,d50在4-6微米之间,可降低粉碎成本。本发明采用水热法制备偶氮金属络合物,使用的反应介质为蒸馏水或去离子水,而未使用任何有机溶剂,反应可以一步到位,无需再加酸或碱分散,工艺路线短,安全可靠,不会对反应、干燥设备产生腐蚀。附图说明图1为实施例1的宽角x射线衍射(wxrd)图图2为对比例1的宽角x射线衍射(wxrd)图图3、图4为实施例1的扫描电镜(sem)图(未经超细气流粉碎)图5、图6为对比例1的扫描电镜(sem)图(经超细气流粉碎)由图1-2可知,水热法与溶剂法相比,所制备的产物晶型基本一致;由图3-6可知,水热法与溶剂法相比,所制备的产物晶体形貌和原晶粒径差别很大,水热法更容易得到原晶粒径纤细的产物。具体实施方式下面通过具体实施例对本发明进行详细说明。实施例1通式ⅰ化合物的结构式为制备上述通式ⅰ偶氮化中间体:盐酸148ml溶解于560g蒸馏水中,加入4-氯-2-氨基苯酚84g,搅拌至完全溶解后冷却至0℃或更低,把溶解在80g蒸馏水中的亚硝酸钠42g缓慢滴入上述的盐酸溶液中,边添加适量冰块保持反应体系温度在-2℃~2℃,滴加完毕后,保持反应在2℃以下反应1.5h,加入氨基磺酸反应10分钟,用淀粉碘化钾试纸确认不再有过剩的亚硝酸,配成重氮液待用;将纯碱12.9g、烧碱47.3g完全溶解于600g蒸馏水中,加入β-萘酚84g,室温下搅拌至溶解完毕,往其中注入上述重氮液,调节并保持ph在8.8,于20℃~22℃反应3h后,用间苯二酚来确认反应终点,然后进行抽滤,所得滤饼以每次500ml蒸馏水洗涤至滤液电导率低于200微西为止,抽滤后所得滤饼在105℃下干燥即得上述结构式的偶氮中间体。将通式ⅰ化合物59.6g、甲酸铬18.7g,加入到705g蒸馏水中,投至反应釜,升温至80℃,在0.05mpa下反应10h,结束反应后,冷却至常温后抽滤,分离出固体物质,以500ml蒸馏水/次洗涤至滤液电导率低于200微西为止,抽滤,滤饼干燥即得偶氮铬络合物,结构式为实施例2通式ⅰ化合物的结构式为:参照实施例1,制备上述结构式的偶氮中间体。将通式ⅰ化合物83.6g、fecl3·6h2o27.02g,加入到443g去离子水中,投至反应釜,升温至100℃,在0.1mpa下反应8h,结束反应后,冷却至常温后抽滤,分离出固体物质,水洗,干燥即得偶氮铁络合物,结构式为实施例3通式ⅰ化合物的结构式为:参照实施例1,制备上述结构式的偶氮中间体。将通式ⅰ化合物83.6g、乳酸铬34.9g,加入到300g蒸馏水中,投至反应釜,升温至120℃,在0.65mpa下反应6h,结束反应后,冷却至常温后抽滤,分离出固体物质,水洗,干燥即得偶氮铬络合物,结构式为实施例4通式ⅰ化合物的结构式为:参照实施例1,制备上述结构式的偶氮中间体。将通式ⅰ化合物79.6g、硫酸铁20.0g,加入到320g蒸馏水中,投至反应釜,升温至130℃,在0.47mpa下反应5h,结束反应后,冷却至常温后抽滤,分离出固体物质,水洗,干燥即得偶氮三价铁络合物,结构式为实施例5通式ⅰ化合物的结构式为:参照实施例1,制备上述结构式的偶氮中间体。将通式ⅰ化合物72.6g、fecl3·6h2o27.02g,加入到93g蒸馏水中,投至反应釜,升温至140℃,在0.76mpa下反应4.5h,结束反应后,冷却至常温后抽滤,分离出固体物质,水洗,干燥即得偶氮铁络合物,结构式为实施例6通式ⅰ化合物的结构式为:参照实施例1,制备上述结构式的偶氮中间体。将通式ⅰ化合物238.4g、氯化铝13.34g,加入到960g蒸馏水中,投至反应釜,升温至150℃,在0.78mpa下反应3h,结束反应后,冷却至常温后抽滤,分离出固体物质,水洗,干燥即得偶氮铝络合物,结构式为实施例7通式ⅰ化合物的结构式为:参照实施例1,制备上述结构式的偶氮中间体。将通式ⅰ化合物59.6g、氢氧化锌9.94g,加入到160g蒸馏水中,投至反应釜,升温至160℃,在0.95mpa下反应4h,结束反应后,冷却至常温后抽滤,分离出固体物质,水洗,干燥即得偶氮锌络合物,结构式为实施例8通式ⅰ化合物的结构式为:参照实施例1,制备上述结构式的偶氮中间体。将通式ⅰ化合物139.6g、醋酸铜18.2g,加入到492g蒸馏水中,投至反应釜,升温至180℃,在3.0mpa下反应4h,结束反应后,冷却至常温后抽滤,分离出固体物质,水洗,干燥即得偶氮铜络合物,结构式为实施例9通式ⅰ化合物的结构式为:参照实施例1,制备上述结构式的偶氮中间体。将通式ⅰ化合物79.6g、硫酸亚铁15.2g,加入到400g蒸馏水中,投至反应釜,升温至190℃,在6.0mpa下反应2h,结束反应后,冷却至常温后抽滤,分离出固体物质,水洗,干燥即得偶氮亚铁络合物,结构式为实施例10通式ⅰ化合物的结构式为:参照实施例1,制备上述结构式的偶氮中间体。将通式ⅰ化合物72.6g、二水合氯化铜17.1g,加入到350g蒸馏水中,投至反应釜,升温至200℃,在7.6mpa下反应1h,结束反应后,冷却至常温后抽滤,分离出固体物质,水洗,干燥即得偶氮铜络合物,结构式为实施例11通式ⅰ化合物的结构式为:参照实施例1,制备上述结构式的偶氮中间体。将通式ⅰ化合物72.6g、氯化锌13.63g,加入到460g蒸馏水中,投至反应釜,升温至230℃,在8.9mpa下反应2.5h,结束反应后,冷却至常温后抽滤,分离出固体物质,水洗,干燥即得偶氮锌络合物,结构式为实施例12通式ⅰ化合物的结构式为:参照实施例1,制备上述结构式的偶氮中间体。将通式ⅰ化合物83.73g、硫酸镍15.5g,加入到410g蒸馏水中,投至反应釜,升温至250℃,在9.6mpa下反应1h,结束反应后,冷却至常温后抽滤,分离出固体物质,水洗,干燥即得偶氮镍络合物,结构式为实施例13通式ⅰ化合物的结构式为:参照实施例1,制备上述结构式的偶氮中间体。将通式ⅰ化合物79.6g、氯化钴13.0g,加入到330g蒸馏水中,投至反应釜,升温至280℃,在10.0mpa下反应0.5h,结束反应后,冷却至常温后抽滤,分离出固体物质,水洗,干燥即得偶氮钴络合物,结构式为实施例14通式ⅰ化合物的结构式为:参照实施例1,制备上述结构式的偶氮中间体。将通式ⅰ化合物72.6g、氯化锰12.6g,加入到390g蒸馏水中,投至反应釜,升温至300℃,在10.0mpa下反应0.5h,结束反应后,冷却至常温后抽滤,分离出固体物质,水洗,干燥即得偶氮锰络合物,结构式为实施例15取实施例1制得的偶氮铬络合物56g,加入到200mlph为8.5的naoh溶液中,60℃下搅拌反应2h,制得抗衡离子为na+的偶氮铬金属络合物。实施例16取实施例9制得的偶氮铁化合物60g,加入200ml的ph为9.0的koh溶液,于50℃下搅拌反应2小时,制得抗衡离子为k+的偶氮铁金属络合物。对比例1通式ⅰ化合物的结构式为:对照实施例1,按照如下方法制备偶氮铬络合物:将59.6g上述化学式的偶氮化合物,和18.7g甲酸铬加入到150g乙二醇和200g乙二醇单乙醚的混合溶剂中,在128℃下搅拌、回流4.5小时后,冷却到90℃,进行反离子调节,其中37%盐酸:蒸馏水=1:2(v/v),络合物与酸化液比(w/v)=1:4,分散1小时后,过滤分离得到固体物,水洗至ph=6,烘干得到比较产物1。比较产物1的宽角x射线衍射图(wxrd)见附件1,经超细气流粉碎后扫描电镜(sem)图见附件2。比较产物1结构式为如下图结构式所示偶氮铬络合物:对比例2通式ⅰ化合物的结构式为:参照对比例1,按照如下方法制备偶氮铁络合物将83.6g上述化学式的偶氮化合物,和20g硫酸铁,加入到150g乙二醇和200g乙二醇单乙醚的混合溶剂中,在128℃下搅拌、回流4.5小时后,冷却到90℃,进行反离子调节,其中37%盐酸:蒸馏水=1:2(v/v),络合物与酸化液比(w/v)=1:4,分散1小时后,过滤分离得到固体物,水洗至ph=6,烘干得到比较产物2。比较产物2结构式为如下图结构式所示偶氮铁络合物:对比例3通式ⅰ化合物的结构式为:参照对比例2,制得如下结构式偶氮铁络合物:将79.6g上述化学式的偶氮化合物,20g硫酸铁,加入到150g乙二醇和200g乙二醇单乙醚混合溶剂中,在125℃下搅拌、回流4.5小时后,冷却到90℃,,进行反离子调节,其中37%盐酸:蒸馏水=1:2(v/v),络合物与酸化液比(w/v)=1:4,分散1小时后,过滤分离得到固体物,水洗至ph=6,烘干得到比较产物3。比较产物3结构式为如下图结构式所示偶氮铁络合物:实施例17检测1、测定实施例1-16及对比例1-3中偶氮金属络合物产物的粒径,计量d50值,测量结果如表1所示。2、将对比例1-3得到的偶氮金属络合物经超细气流粉碎达到与实施例1-16的粒径基本一致时,它们的起电性能测试结果见表2所示,方法如下:精确(0.0002g)称取正电性纯载体2.4g于5ml称量瓶中,分别称取0.1克实施例1-14产物和粉碎后对比例1-3产物样品置于称量瓶中,混合摇匀10min,准确(0.0002g)称取均匀的混合物0.2克,小心投入带电量测试仪的法拉第电筒中心部位,密闭法拉第电筒,数据归零,用0.10mpa的氮气吹气30秒,记录电性和电量数据。实施例18将色调剂专用树脂94g、炭黑4.1g、石蜡1g,实施例1合成的偶氮铬金属络合剂电荷控制剂0.8g。加热熔融混炼,冷却后经粉碎分级,收集8-10微米颗粒为色调剂母体;按母体重量的0.8%混入疏水纳米二氧化硅,充分混合得到色调剂。参照实施例18,制备不同的色调剂,除了电荷控制剂采用不同的品种和用量,其他部分不变,详细如下:实施例19采用的0.8g电荷控制剂来自实施例2合成的产物。实施例20采用的0.8g电荷控制剂来自实施例3合成的产物。实施例21采用的0.8g电荷控制剂来自实施例4合成的产物。实施例22采用的0.8g电荷控制剂来自实施例5合成的产物。实施例23采用的0.8g电荷控制剂来自实施例6合成的产物。实施例24采用的0.8g电荷控制剂来自实施例7合成的产物。实施例25采用的0.8g电荷控制剂来自实施例8合成的产物。实施例26采用的0.8g电荷控制剂来自实施例9合成的产物。实施例27采用的0.8g电荷控制剂来自实施例10合成的产物。实施例28采用的0.8g电荷控制剂来自实施例11合成的产物。实施例29采用的0.8g电荷控制剂来自实施例12合成的产物。实施例30采用的0.8g电荷控制剂来自实施例13合成的产物。实施例31采用的0.8g电荷控制剂来自实施例14合成的产物。实施例32采用的0.8g电荷控制剂来自实施例15合成的产物。实施例33采用的0.8g电荷控制剂来自实施例16合成的产物。实施例34采用的0.8g电荷控制剂来自比较对比例1合成的产物。实施例35采用的0.8g电荷控制剂来自比较对比例2合成的产物。实施例36采用的0.8g电荷控制剂来自比较对比例3合成的产物。实施例18-36的色调剂电性能测量结果见表2。方法如下:精确(0.0002g)称取正电性纯载体2.4g于5ml称量瓶中,分别称取0.1克实施例15-31色调剂样品置于称量瓶中,混合摇匀10min,准确(0.0002g)称取均匀的混合物0.2克,小心投入带电量测试仪的法拉第电筒中心部位,密闭法拉第电筒,数据归零,用0.10mpa的氮气吹气1min,记录电性和电量数据。表1不同偶氮金属络合物原生粒径的d50/μm偶氮金属络合物例子原生粒径(d50/μm)实施例14.92实施例25.24实施例36.20实施例45.48实施例54.67实施例65.38实施例76.02实施例85.72实施例94.61实施例106.10实施例114.95实施例124.86实施例135.02实施例146.17实施例154.98实施例165.02对比例126.80对比例228.70对比例330.60由表1结果可见,水热法制备偶氮金属络合物,其原生粒径的d50比传统有机溶剂发制备的要小很多。表2实施例1-16以及对比例1-3的产物和它们制成的色调剂电性能测试结果结论:从表2数据可见,本发明的所提供的水热法制备的偶氮金属络合物电荷调节剂具有实用的带电量。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 金属络合物及使用了其的发光元件的制作方法与工艺
- 一种金属杂化POSS络合物及其制备方法和应用与流程
- 新的手性金属络合物及其用于通过1H NMR光谱分析带电化合物的手性的用途的制作方法与工艺
- 新的手性金属络合物及其用于通过1H NMR光谱分析带电化合物的手性的用途的制作方法与工艺
- 金属络合物催化剂和采用金属络合物催化剂的聚合方法与流程
- 金属络合物的合成及其用途的制作方法与工艺
- 是SOD模拟物的金属络合物作为食品试剂和作为化妆品的应用的制作方法与工艺
- 还原球团中金属铁含量的测定方法与流程
- 具有三脚架配位体的过渡金属络合物和其在OLED中的用途的制作方法与工艺
- 在金(I)络合物存在的情况下基于炔烃与二甲基呋喃的分子间反应形成色满的制造方法与工艺
- 一种金属杂化POSS络合物及其制备方法和应用与流程
- 新的手性金属络合物及其用于通过1H NMR光谱分析带电化合物的手性的用途的制作方法与工艺
- 新的手性金属络合物及其用于通过1H NMR光谱分析带电化合物的手性的用途的制作方法与工艺
- 金属络合物催化剂和采用金属络合物催化剂的聚合方法与流程
- 金属络合物的合成及其用途的制作方法与工艺
- 是SOD模拟物的金属络合物作为食品试剂和作为化妆品的应用的制作方法与工艺
- 具有三脚架配位体的过渡金属络合物和其在OLED中的用途的制作方法与工艺
- 在金(I)络合物存在的情况下基于炔烃与二甲基呋喃的分子间反应形成色满的制造方法与工艺
- 金属络合物制剂的制造方法与工艺
- 金属络合物和使用该金属络合物的发光元件的制造方法与工艺