含咔唑树枝蓝光热激发延迟荧光芳香材料及其暖白光器件制备方法和应用与流程
本发明涉及热激发延迟荧光材料和制备方法及应用。
背景技术:
制备白色的有机发光器件在固态照明和全色显示上有很大的意义。蓝光和黄光构建的白光器件中,纯色的蓝光和黄光的发光效率对整个器件起到重要的影响。所以说构建出高效的蓝光材料有利于提高白光器件的性能。以报道的白光或暖白光器件大多有三个或两个发光层构成,设备要求高,制作工艺复杂。已报到的单层白光器件效率也较低,如lianduan报道的单层白光器件效率在10%左右。
膦光有机电致发光二极管在第一代荧光的有机发光二极管(organiclight-emittingdiodes,oled)之后取得了较大的成功。含贵金属的有机金属膦光体由于有效的自旋轨道耦合而具有发射三重态,能够实现更高得的外量子效率(externalquantumefficiency,eqe)。尽管报道了高效的蓝色膦光有机发光二极管,但是使用昂贵和不可再生的贵金属,妨碍了它们的普及。目前引人引人关注的是,具有小的单重态-三重态能极差的热激活延迟荧光(thermallyactivateddelayedfluorescence,tadf)材料,它可以将三重态激子上转换成单线态激发态,从而发射荧光,理论上可获得100%的外量子效率。
技术实现要素:
本发明的目的是为了解决现有tadf蓝光客体材料单一性差,蓝光色纯度低和效率低的问题,而提供含咔唑树枝蓝光热激发延迟荧光芳香材料及其暖白光器件制备方法和应用。
含咔唑树枝蓝光热激发延迟荧光芳香材料的结构式为
其中,所述的r为h或t-c4h9。
含咔唑树枝蓝光热激发延迟荧光芳香材料由含氮杂环化合物树枝和二溴二氟苯制备而成。
含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中。
含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在热激发延迟荧光电致发光器件中。
含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在磷光器件中。
含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在暖白光器件中。
本发明的优点:
一、tadf客体材料分子结构主要由电子给体和电子受体构成。电子给体和受体形成电子推拉结构,使分子扭曲,以平衡电荷的传输。热激发延迟荧光客体材料用于oled时,需要分散在主体中以防止因其三线态-三线态激子湮灭(tta)引起的能量损失。当分子的给体或受体基团有较大的空间位阻时,不仅可以使tadf分子的刚性增强,还可以防止分子间的湮灭,二苯基膦氧具有适当的吸电子能力,可以使光谱的发射峰在蓝光区域,采用咔唑树枝作为电子给体使分子的刚性增强,减少了由于分子形变产生的能量损失。膦氧和咔唑基团的结合使分子具有高的三线态能,和良好的载流子传输能力,所以将其用于热激发延迟荧光材料和黄色膦光材料的主体,黄光磷光材料以较低的浓度掺杂在蓝色tadf材料中,可以减少tadf材料的tta机率。另外较低的掺杂浓度降低了磷光材料的使用量,减少了器件成本;
二、二苯基膦氧具有适当的吸电子能力,可以使光谱的发射峰在蓝光区域,采用咔唑树枝作为电子给体使分子的刚性增强,减少了由于分子形变产生的能量损失。膦氧和咔唑基团的结合使分子具有高的三线态能,和良好的载流子传输能力,所以将其用于热激发延迟荧光材料和黄色膦光材料的主体。并在此基础上构建了高效的白光器件;
三、本发明制备的含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在磷光器件中,制备的磷光器件的半峰宽为65nm~72nm;
四、取失重5%为裂解温度,本发明制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的裂解温度为413℃~445℃;
五、本发明制备的含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中,制备的热激发延迟荧光电致发光器件的电流密度为:62ma/cm2~93ma/cm2,亮度为1641cd/m2~5572cd/m2,电流效率分别为2.7ma/cm2~18.7ma/cm2,功率效率为3.5lm/w~9.7lm/w,外量子效率为3.0%~12.2%;
六、本发明制备的含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在热激发延迟荧光电致发光器件中,制备的热激发延迟荧光电致发光器件的的亮度-外量子效率分别为10.2%~13.8%,发光光谱分别为472nm~488nm;
七、本发明制备的含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在膦光器件中,制备的膦光器件的外量子效率分别为7%~19.0%,光谱均在565nm;
八、本发明制备的含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在暖白光器件白光器件的外量子效率为15.3%,光谱为488nm和556nm。
本发明可获得含咔唑树枝蓝光热激发延迟荧光芳香材料的方法。
附图说明
图1为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料溶于二氯甲烷中的光谱曲线,图1中“■”为紫外吸收光谱曲线,“▲”为荧光发射光谱曲线;
图2为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的热重分析谱图;
图3为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料溶于二氯甲烷中的光谱曲线,图1中“■”为紫外吸收光谱曲线,“▲”为荧光发射光谱曲线;
图4为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的热重分析谱图;
图5为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料溶于二氯甲烷中的光谱曲线,图1中“■”为紫外吸收光谱曲线,“▲”为荧光发射光谱曲线;
图6为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的热重分析谱图;
图7为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料溶于二氯甲烷中的光谱曲线,图1中“■”为紫外吸收光谱曲线,“▲”为荧光发射光谱曲线;
图8为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的热重分析谱图;
图9为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料溶于二氯甲烷中的光谱曲线,图1中“■”为紫外吸收光谱曲线,“▲”为荧光发射光谱曲线;
图10为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的热重分析谱图;
图11为实施例六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料溶于二氯甲烷中的光谱曲线,图1中“■”为紫外吸收光谱曲线,“▲”为荧光发射光谱曲线;
图12为实施例六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的热重分析谱图;
图13为含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的电压-电流密度关系曲线,图13中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
图14为含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的电压-亮度关系曲线,图14中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
图15为含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的亮度-电流效率关系曲线,图15中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
图16为含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的亮度-功率效率关系曲线,图16中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
图17为含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的亮度-外量子效率关系曲线,图17中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
图18为含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的电致发光光谱,图18中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
图19为含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的亮度-外量子效率关系曲线,图19中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
图20为含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的电致发光光谱,图20中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
图21为含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在磷光器件中的亮度-外量子效率关系曲线,图21中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
图22为含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在磷光器件中的电致发光光谱,图22中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
图23为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在暖白光器件中的亮度-外量子效率关系曲线;
图24为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在暖白光器件中的电致发光光谱。
具体实施方式
具体实施方式一:本实施方式是含咔唑树枝蓝光热激发延迟荧光芳香材料的结构式为
具体实施方式二:本实施方式是含咔唑树枝蓝光热激发延迟荧光芳香材料由含氮杂环化合物树枝和二溴二氟苯制备而成。
具体实施方式三:本实施方式与具体实施方式一的不同点是:所述的含氮杂环化合物树枝具体是按以下步骤完成的:
一、制备咔唑保护:在冰水浴和搅拌的条件下,向二甲基甲酰胺中加入咔唑,再加入氢氧化钾,再在冰水浴和搅拌的条件下反应1.5h~2.5h,再加入对甲基苯磺酰氯溶液,再在冰水浴和搅拌的条件下反应3h~5h,反应结束后,使用水和二氯甲烷的混合液进行萃取,得到二氯甲烷层,然后使用无水硫酸钠对二氯甲烷层进行干燥,再使用旋转蒸发仪在常压下蒸发出二氯甲烷,再冷却至室温,得到粗反应产物ⅰ;向粗反应产物ⅰ中加入乙酸乙酯析出晶体,再进行抽滤,再使用无水乙醇对析出的晶体清洗3次~5次,再在室温条件下干燥,得到白色晶体,即为咔唑保护;
步骤一中所述的咔唑的物质的量与二甲基甲酰胺的体积比为(5mmol~8mmol):15ml;
步骤一中所述的氢氧化钾的物质的量与二甲基甲酰胺的体积比为(20mmol~25mmol):15ml;
步骤一中所述的对甲基苯磺酰氯溶液为对甲基苯磺酰氯溶解到二甲基甲酰胺得到,对甲基苯磺酰氯溶液中对甲基苯磺酰氯的物质的量与二甲基甲酰胺的体积比为(5mmol~8mmol):5ml;
步骤一中所述的对甲基苯磺酰氯溶液与二甲基甲酰胺的体积比为(4ml~6ml):15ml;
步骤一中所述的水和二氯甲烷的混合液中水和二氯甲烷的体积比为1:5;
步骤一中所述的咔唑保护的结构式为
二、制备3,6-双碘保护咔唑:将咔唑保护、碘加入到冰乙酸中,再在温度为120℃~130℃下加热回流20min~40min,再以20滴/min~40滴/min的滴加速度滴入质量分数为70%~85%的浓硝酸,再在温度为120℃~130℃下反应2h~4h,再自然冷却至室温,再进行抽滤,使用无水乙醇对抽滤后得到的固体物质洗涤3次~5次,得到白色絮状固体,即为3,6-双碘保护咔唑;
步骤二中所述的咔唑保护的物质的量与冰乙酸的体积比为(8mmol~12mmol):40ml;
步骤二中所述的碘的物质的量与冰乙酸的体积比为(10mmol~12mmol):40ml;
步骤二中所述的质量分数为70%~85%的浓硝酸与冰乙酸的体积比为(13~18):40;
步骤二中所述的3,6-双碘保护咔唑的结构式为
三、制备含氮杂环化合物保护咔唑:将3,6-双碘保护咔唑、含氮杂环化合物、18冠6相转移催化剂加入到干燥的三颈瓶中混合均匀,再在氮气气氛保护下滴加硝基苯,搅拌均匀,再在氩气气氛和温度为150℃~170℃下油浴加热反应45h~50h,得到反应液;使用水和二氯甲烷的混合液对反应液进行萃取,得到二氯甲烷层,然后使用无水硫酸钠对二氯甲烷层进行干燥,再使用旋转蒸发仪在常压下蒸发出二氯甲烷,得到粗反应产物ⅱ,最后以溶剂ⅰ为淋洗剂,将粗反应产物ⅱ经柱层析纯化,得到反应产物ⅱ,即为含氮杂环化合物保护咔唑;
步骤三中所述的含氮杂环化合物为咔唑或3,6-二叔丁基咔唑;
步骤三中所述的3,6-双碘保护咔唑的物质的量与硝基苯的体积比为1mmol:(10ml~20ml);
步骤三中所述的3,6-双碘保护咔唑与含氮杂环化合物的物质的量1:(2~2.5);
步骤三中所述的3,6-双碘保护咔唑与18冠6相转移催化剂的物质的量1:(0.02~0.03);
步骤三中所述的溶剂ⅰ为二氯甲烷和石油醚的混合液,二氯甲烷和石油醚的体积比为1:3;
步骤三中所述的水和二氯甲烷的混合液中水和二氯甲烷的体积比为1:5;
步骤三中所述的含氮杂环化合物保护咔唑的结构式为
四、制备含氮杂环化合物树枝:将含氮杂环化合物保护咔唑、氢氧化钾、1,4-二氧六环和蒸馏水混合,再在温度为110℃~130℃和搅拌的条件下反应10h~12h,再使用水来淬灭反应,得到含有反应物的混合溶液;使用二氯甲烷对含有反应物的混合液ⅱ进行萃取,使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅲ;以溶剂ⅱ为淋洗剂,将粗反应产物ⅲ经柱层析纯化,得到反应产物ⅲ,即为含氮杂环化合物树枝;
步骤四中所述的溶剂ⅱ为二氯甲烷和石油醚的混合溶液,二氯甲烷和石油醚的体积比为3:2;
步骤四中所述的含氮杂环化合物保护咔唑的物质的量与蒸馏水的体积比为(0.1mmol~0.2mmol):20ml;
步骤四中所述的氢氧化钾的物质的量与蒸馏水的体积比为(0.5mmol~0.75mmol):20ml;
步骤四中所述的1,4-二氧六环与蒸馏水的体积比为(1.5~2):20;
步骤四中所述的含氮杂环化合物树枝的结构式为
具体实施方式四:本实施方式与具体实施方式一的不同点是
一、制备含氮杂环化合物树枝取代的二溴苯中间体:将1,4-二溴-2,5-二氟苯、含氮杂环化合物树枝、碳酸钾加入到二甲基亚砜中,再在氩气气氛和温度为120℃~140℃下反应4h~8h,得到含有反应产物的混合溶液;使用水和二氯甲烷的混合液对含有反应物的混合液ⅲ对含有反应产物的混合溶液进行萃取,使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅳ;使用溶剂ⅲ为淋洗剂,将粗反应产物ⅳ经柱层析纯化,得到反应产物ⅳ,即为含氮杂环化合物树枝取代的二溴苯中间体;
步骤一中所述的1,4-二溴-2,5-二氟苯的物质的量与二甲基亚砜的体积比为(0.8mmol~1.2mmol):5ml;
步骤一中所述的含氮杂环化合物树枝的物质的量与二甲基亚砜的体积比为(2mmol~2.5mmol):5ml;
步骤一中所述的碳酸钾的物质的量与二甲基亚砜的体积比为(2mmol~2.5mmol):5ml;
步骤一中所述的水和二氯甲烷的混合液中水和二氯甲烷的体积比为5:1;
步骤一中所述的溶剂ⅲ为石油醚和二氯甲烷的混合液,石油醚和二氯甲烷的体积比为8:1;
步骤一中所述的含氮杂环化合物树枝取代的二溴苯中间体的结构式为
二、在-100℃~0℃下将含氮杂环化合物树枝取代的二溴苯中间体和四氢呋喃混合,再在搅拌条件下滴加正丁基锂,再在温度为-100℃下搅拌反应10h~12h,再在搅拌的条件下滴加二苯基氯化膦,再在搅拌速度为60r/min~80r/min下磁力搅拌5h~12h,再使用水来淬灭反应,再使用水猝灭反应,再加入质量分数为30%的双氧水,再在搅拌速度为60r/min~80r/min下搅拌0.5h~1.5h,得到含有反应物的混合液ⅴ;使用二氯甲烷对含有反应物的混合液ⅴ进行萃取,然后使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物;以溶剂ⅴ为淋洗剂,将粗反应产物经柱层析纯化,再使用乙酸乙酯进行重结晶,得到含咔唑树枝蓝光热激发延迟荧光芳香材料;
步骤二中所述的含氮杂环化合物树枝取代的二溴苯中间体的物质的量与四氢呋喃的体积比为(1mmol~5mmol):(5ml~20ml);
步骤二中所述的正丁基锂的物质的量与四氢呋喃的体积比为(1mmol~5mmol):(5ml~20ml);
步骤二中所述的二苯基氯化膦的物质的量与四氢呋喃的体积比为(1mmol~5mmol):(5ml~20ml);
步骤二中所述的质量分数为30%的双氧水的物质的量与四氢呋喃的体积比为(1mmol~20mmol):(5ml~20ml);
步骤二中所述的溶剂ⅴ为石油醚和甲醇的混合液,石油醚和甲醇的体积比为50:1。其他与具体实施方式一相同。
具体实施方式五:本实施方式与具体实施方式一的不同点是:
一、将1,5-二溴-2,4-二氟苯、含氮杂环化合物树枝和碳酸钾加入到二甲基亚砜中,再在氩气气氛和温度为120℃~160℃下反应6h~12h,得到含有反应物的混合液ⅰ;使用水和二氯甲烷的混合液对含有反应物的混合液ⅰ进行萃取,然后使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅰ;以溶剂ⅰ为淋洗剂,将粗反应产物ⅰ经柱层析纯化,得到反应产物ⅰ,即为含氮杂环化合物树枝取代的二溴苯中间体;
步骤一中所述的溶剂ⅰ为石油醚和二氯甲烷的混合溶液,溶剂ⅰ中石油醚与二氯甲烷的体积比为5:1;
步骤一中所述的1,5-二溴-2,4-二氟苯的物质的量与二甲基亚砜的体积比为(0.8mmol~1.2mmol):5ml;
步骤一中所述的含氮杂环化合物树枝的物质的量与二甲基亚砜的体积比为(1.8mmol~2.5mmol):5ml;
步骤一中所述的碳酸钾的物质的量与二甲基亚砜的体积比为(1.8mmol~2.5mmol):5ml;
步骤一中所述的水和二氯甲烷的混合液中水和二氯甲烷的体积比为1:5;
步骤一中所述的含氮杂环化合物树枝取代的二溴苯中间体的结构式为
二、在-100℃~0℃下将含氮杂环化合物树枝取代的二溴苯中间体和四氢呋喃混合,再在搅拌条件下滴加正丁基锂,再在温度为-100℃~0℃下搅拌反应10h~36h,再在搅拌的条件下滴加二苯基氯化膦,再在搅拌速度为60r/min~80r/min下磁力搅拌5h~24h,再使用水来淬灭反应,再使用水猝灭反应,再加入质量分数为30%的双氧水,再在搅拌速度为60r/min~80r/min下搅拌0.5h~1.5h,得到含有反应物的混合液ⅱ;使用二氯甲烷对含有反应物的混合液ⅱ进行萃取,然后使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅱ;以溶剂ⅱ为淋洗剂,将粗反应产物ⅱ经柱层析纯化,再使用乙酸乙酯进行重结晶,得到含咔唑树枝蓝光热激发延迟荧光芳香材料;
步骤二中所述的含氮杂环化合物树枝取代的二溴苯中间体的物质的量与四氢呋喃的体积比为2mmol:(5ml~20ml);
步骤二中所述的正丁基锂的物质的量与四氢呋喃的体积比为(1mmol~5mmol):(5ml~20ml);
步骤二中所述的二苯基氯化膦的物质的量与四氢呋喃的体积比为(1mmol~5mmol):(5ml~20ml);
步骤二中所述的溶剂ⅱ为石油醚和甲醇的混合物,溶剂ⅱ中石油醚合甲醇的体积比为20:1;
步骤二中所述的双氧水的物质的量与四氢呋喃的体积比为(1mmol~20mmol):(5ml~20ml)。
具体实施方式六:本实施方式与具体实施方式一的不同点是:
一、将1,2-二溴-4,5-二氟苯、含氮杂环化合物树枝和碳酸钾加入到二甲基亚砜中,再在氩气气氛和温度为120℃~160℃下反应6h~12h,得到含有反应物的混合液ⅰ;使用水和二氯甲烷的混合溶液对含有反应物的混合液ⅰ进行萃取,然后使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅰ;以溶剂ⅰ为淋洗剂,将粗反应产物ⅰ经柱层析纯化,得到反应产物ⅰ;
步骤一中所述的1,2-二溴-4,5-二氟苯的物质的量与二甲基亚砜的体积比为(0.8mmol~1.2mmol):5ml;
步骤一中所述的含氮杂环化合物树枝的物质的量与二甲基亚砜的体积比为(1.8mmol~2.5mmol):5ml;
步骤一中所述的碳酸钾的物质的量与二甲基亚砜的体积比为(1.8mmol~2.5mmol):5ml;
步骤一中所述的溶剂ⅰ为石油醚和二氯甲烷的混合液,溶剂ⅰ中石油醚和二氯甲烷的体积比为5:1;
步骤一中所述的水和二氯甲烷的混合溶液中水和二氯甲烷的体积比为1:5;
步骤一中所述的反应产物ⅰ的结构式为
二、在-100℃~0℃下将反应产物ⅰ、四氢呋喃和乙醚混合,再在搅拌条件下滴加正丁基锂,再在温度为-100℃~0℃下搅拌反应10h~36h,再在搅拌的条件下滴加二苯基氯化膦,再在搅拌速度为60r/min~80r/min下磁力搅拌5h~24h,再使用水来淬灭反应,得到含有反应物的混合液ⅱ;使用二氯甲烷对含有反应物的混合液ⅱ进行萃取,使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅱ;以溶剂ⅱ为淋洗剂,将粗反应产物ⅱ经柱层析纯化,再使用乙酸乙酯进行重结晶,得到反应产物ⅱ;
步骤二中所述的反应产物ⅰ的物质的量与四氢呋喃的体积比为2mmol:(5ml~20ml);
步骤二中所述的四氢呋喃与乙醚的体积比为1:1;
步骤二中所述的正丁基锂的物质的量与四氢呋喃的体积比为(1mmol~5mmol):(5ml~20ml);
步骤二中所述的二苯基氯化膦的物质的量与四氢呋喃的体积比为(1mmol~5mmol):(5ml~20ml);
步骤二中所述的溶剂ⅱ为石油醚和二氯甲烷的混合溶液,溶剂ⅱ中石油醚和二氯甲烷的体积比为5:1;
步骤二中所述的反应产物ⅱ的结构式为
三、在-100℃~0℃下将反应产物ⅱ和四氢呋喃混合,再在搅拌的条件下滴加正丁基锂,再在温度为-100℃~0℃下搅拌反应10h~36h,再在搅拌的条件下滴加二苯基氯化膦,再在搅拌速度为60r/min~80r/min下磁力搅拌5h~24h,再使用水来淬灭反应,再加入质量分数为30%的双氧水,再在搅拌速度为60r/min~80r/min下搅拌0.5h~1.5h,得到含有反应物的混合液ⅲ;使用二氯甲烷对含有反应物的混合液ⅲ进行萃取,然后使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅲ;以溶剂ⅲ为淋洗剂,将粗反应产物ⅲ经柱层析纯化,再使用乙酸乙酯进行重结晶,得到含咔唑树枝蓝光热激发延迟荧光芳香材料;
步骤三中所述的反应产物ⅱ的物质的量与四氢呋喃的体积比为2mmol:(5ml~20ml);
步骤三中所述的正丁基锂的物质的量与四氢呋喃的体积比为(1mmol~5mmol):(5ml~20ml);
步骤三中所述的二苯基氯化膦的物质的量与四氢呋喃的体积比为(1mmol~5mmol):(5ml~20ml);
步骤三中所述的溶剂ⅲ为石油醚和二氯甲烷的混合溶液,溶剂ⅲ中石油醚和二氯甲烷的体积比为5:1;
步骤三中所述的双氧水的物质的量与四氢呋喃的体积比为(1mmol~20mmol):(5ml~20ml)。其他与具体实施方式一相同。
具体实施方式七:本实施方式与具体实施方式一的不同点是:含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中,热激发延迟荧光电致发光器件具体是按以下方法制备的:
一、将依次使用丙酮、无水乙醇和去离子水各清洗3次~5次的ito基片安装到旋转蒸发仪上,再旋涂上pedot:pss,再在温度为120℃下烘烤8min~12min,得到表面含有厚度为50nmpedot:pss薄膜的ito基片;
二、在表面含有厚度为50nmpedot:pss薄膜的ito基片上旋涂发光材料溶液,再在温度为100℃下烘烤8min~12min,得到厚度为70nm的发光层;
步骤二中所述的涂发光材料溶液由发光材料和氯苯混合而成;所述的涂发光材料溶液中发光材料的质量与氯苯的体积比为10mg:1ml;所述的发光材料为dpetpo和含咔唑树枝蓝光热激发延迟荧光芳香材料组成,且dpetpo与含咔唑树枝蓝光热激发延迟荧光芳香材料的摩尔比为3:1;
三、在真空度为1×10-6mbar和蒸镀速率为0.1nms-1~0.3nms-1的条件下在发光层上蒸镀tmpypb材料,在发光层上得到厚度为40nm的电子传输层;
四、在真空度为1×10-6mbar和蒸镀速率为0.1nms-1~0.3nms-1的条件下在电子传输层上蒸镀lif材料,在电子传输层上得到厚度为0.5nm的电子注入层;
五、在真空度为1×10-6mbar和蒸镀速率为0.1nms-1~0.3nms-1的条件下在电子注入层上蒸镀金属al材料,在电子注入层上得到厚度为150nm的阴极导电层,再进行封装,得到热激发延迟荧光电致发光器件。
具体实施方式八:本实施方式与具体实施方式一的不同点是:含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在热激发延迟荧光电致发光器件中,热激发延迟荧光电致发光器件具体是按以下方法制备的:
一、将依次使用丙酮、无水乙醇和去离子水各清洗3次~5次的ito基片安装到旋转蒸发仪上,再旋涂上pedot:pss,再在温度为120℃下烘烤8min~12min,得到表面含有厚度为50nmpedot:pss薄膜的ito基片;
二、在表面含有厚度为50nmpedot:pss薄膜的ito基片上旋涂发光材料溶液,再在温度为100℃下烘烤8min~12min,得到厚度为20nm的发光层;
步骤二中所述的涂发光材料溶液由发光材料和氯苯混合而成;所述的涂发光材料溶液中发光材料的质量与氯苯的体积比为10mg:1ml;所述的发光材料为含咔唑树枝蓝光热激发延迟荧光芳香材料和dmac-dps组成,且含咔唑树枝蓝光热激发延迟荧光芳香材料与dmac-dps的摩尔比为8:1;
三、在真空度为1×10-6mbar和蒸镀速率为0.1nms-1~0.3nms-1的条件下在发光层上蒸镀tmpypb材料,在发光层上得到厚度为40nm的电子传输层;
四、在真空度为1×10-6mbar和蒸镀速率为0.1nms-1~0.3nms-1的条件下在电子传输层上蒸镀lif材料,在电子传输层上得到厚度为0.5nm的电子注入层;
五、在真空度为1×10-6mbar和蒸镀速率为0.1nms-1~0.3nms-1的条件下在电子注入层上蒸镀金属al材料,在电子注入层上得到厚度为150nm的阴极导电层,再进行封装,得到热激发延迟荧光电致发光器件。
具体实施方式九:本实施方式与具体实施方式一的不同点是:含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在磷光器件中,磷光器件具体是按以下方法制备的:
一、将依次使用丙酮、无水乙醇和去离子水各清洗3次~5次的ito基片安装到旋转蒸发仪上,再旋涂上pedot:pss,再在温度为120℃下烘烤8min~12min,得到表面含有厚度为50nmpedot:pss薄膜的ito基片;
二、在表面含有厚度为50nmpedot:pss薄膜的ito基片上旋涂发光材料溶液,再在温度为100℃下烘烤8min~12min,得到厚度为20nm的发光层;
步骤二中所述的涂发光材料溶液由发光材料和氯苯混合而成;所述的涂发光材料溶液中发光材料的质量与氯苯的体积比为10mg:1ml;所述的发光材料为含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为10:1;
三、在真空度为1×10-6mbar和蒸镀速率为0.1nms-1~0.3nms-1的条件下在发光层上蒸镀tmpypb材料,在发光层上得到厚度为40nm的电子传输层;
四、在真空度为1×10-6mbar和蒸镀速率为0.1nms-1~0.3nms-1的条件下在电子传输层上蒸镀lif材料,在电子传输层上得到厚度为0.5nm的电子注入层;
五、在真空度为1×10-6mbar和蒸镀速率为0.1nms-1~0.3nms-1的条件下在电子注入层上蒸镀金属al材料,在电子注入层上得到厚度为150nm的阴极导电层,再进行封装,得到热激发延迟荧光电致发光器件。
本实施方式所述的po-01为乙酰丙酮酸二(4-苯基-噻吩[3,2-c]吡啶-c2,n)合铱(iii),购买自西安宝莱光电科技有限公司。
具体实施方式十:本实施方式与具体实施方式一的不同点是:含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在暖白光器件中,暖白光器件的制备方法具体是按以下方法制备的:
一、将依次使用丙酮、无水乙醇和去离子水各清洗3次~5次的ito基片安装到旋转蒸发仪上,再旋涂上pedot:pss,再在温度为120℃下烘烤8min~12min,得到表面含有厚度为50nmpedot:pss薄膜的ito基片;
二、在表面含有厚度为50nmpedot:pss薄膜的ito基片上旋涂发光材料溶液,再在温度为100℃下烘烤8min~12min,得到厚度为20nm的发光层;
步骤二中所述的涂发光材料溶液由发光材料和氯苯混合而成;所述的涂发光材料溶液中发光材料的质量与氯苯的体积比为10mg:1ml;所述的发光材料为含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为30:1;
三、在真空度为1×10-6mbar和蒸镀速率为0.1nms-1~0.3nms-1的条件下在发光层上蒸镀tmpypb材料,在发光层上得到厚度为40nm的电子传输层;
四、在真空度为1×10-6mbar和蒸镀速率为0.1nms-1~0.3nms-1的条件下在电子传输层上蒸镀lif材料,在电子传输层上得到厚度为0.5nm的电子注入层;
五、在真空度为1×10-6mbar和蒸镀速率为0.1nms-1~0.3nms-1的条件下在电子注入层上蒸镀金属al材料,在电子注入层上得到厚度为150nm的阴极导电层,再进行封装,得到热激发延迟荧光电致发光器件。
本实施方式所述的po-01为乙酰丙酮酸二(4-苯基-噻吩[3,2-c]吡啶-c2,n)合铱(iii),购买自西安宝莱光电科技有限公司。
采用以下实施例验证本发明的有益效果:
实施例一:含咔唑树枝蓝光热激发延迟荧光芳香材料具体是按以下步骤制备的:
一、制备咔唑保护:在冰水浴和搅拌的条件下,向15ml二甲基甲酰胺中加入6mmol咔唑,再加入23mmol氢氧化钾,再在冰水浴和搅拌的条件下反应2h,再加入5ml对甲基苯磺酰氯溶液,再在冰水浴和搅拌的条件下反应4h,反应结束后,使用水和二氯甲烷的混合液进行萃取,得到二氯甲烷层,然后使用无水硫酸钠对二氯甲烷层进行干燥,再使用旋转蒸发仪在常压下蒸发出二氯甲烷,再冷却至室温,得到粗反应产物ⅰ;向粗反应产物ⅰ中加入乙酸乙酯析出晶体,再进行抽滤,再使用无水乙醇对析出的晶体清洗3次,再在室温条件下干燥,得到白色晶体,即为咔唑保护;
步骤一中所述的对甲基苯磺酰氯溶液为对甲基苯磺酰氯溶解到二甲基甲酰胺得到,对甲基苯磺酰氯溶液中对甲基苯磺酰氯的物质的量与二甲基甲酰胺的体积比为6mmol:5ml;
步骤一中所述的水和二氯甲烷的混合液中水和二氯甲烷的体积比为1:5;
步骤一中所述的咔唑保护的结构式为
二、制备3,6-双碘保护咔唑:将10mmol咔唑保护、11mmol碘加入到40ml冰乙酸中,再在温度为125℃下加热回流30min,再以30滴/min的滴加速度滴入15ml质量分数为70%的浓硝酸,再在温度为125℃下反应3h,再自然冷却至室温,再进行抽滤,使用无水乙醇对抽滤后得到的固体物质洗涤3次,得到白色絮状固体,即为3,6-双碘保护咔唑;
步骤二中所述的3,6-双碘保护咔唑的结构式为
三、制备含氮杂环化合物保护咔唑:将1mmol3,6-双碘保护咔唑、2.2mmol含氮杂环化合物、0.02mmol18冠6相转移催化剂加入到50ml干燥的三颈瓶中混合均匀,再在氮气气氛保护下滴加15ml硝基苯,搅拌均匀,再在氩气气氛和温度为160℃下油浴加热反应48h,得到反应液;使用水和二氯甲烷的混合液对反应液进行萃取,得到二氯甲烷层,然后使用无水硫酸钠对二氯甲烷层进行干燥,再使用旋转蒸发仪在常压下蒸发出二氯甲烷,得到粗反应产物ⅱ,最后以溶剂ⅰ为淋洗剂,将粗反应产物ⅱ经柱层析纯化,得到反应产物ⅱ,即为含氮杂环化合物保护咔唑;
步骤三中所述的含氮杂环化合物为咔唑;
步骤三中所述的溶剂ⅰ为二氯甲烷和石油醚的混合液,二氯甲烷和石油醚的体积比为1:3;
步骤三中所述的水和二氯甲烷的混合液中水和二氯甲烷的体积比为1:5;
步骤三中所述的含氮杂环化合物保护咔唑的结构式为
四、制备含氮杂环化合物树枝:将0.1mmol含氮杂环化合物保护咔唑、0.65mmol氢氧化钾、1.7ml1,4-二氧六环和20ml蒸馏水混合,再在温度为120℃和搅拌的条件下反应10h,再使用水来淬灭反应,得到含有反应物的混合溶液;使用二氯甲烷对含有反应物的混合液ⅱ进行萃取,使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅲ;以溶剂ⅱ为淋洗剂,将粗反应产物ⅲ经柱层析纯化,得到反应产物ⅲ,即为含氮杂环化合物树枝;
步骤四中所述的溶剂ⅱ为二氯甲烷和石油醚的混合溶液,二氯甲烷和石油醚的体积比为3:2;
步骤四中所述的含氮杂环化合物树枝的结构式为
五、制备含氮杂环化合物树枝取代的二溴苯中间体:将1mmol1,4-二溴-2,5-二氟苯、2.2mmol含氮杂环化合物树枝、2.27mmol碳酸钾加入到5ml二甲基亚砜中,再在氩气气氛和温度为130℃下反应6h,得到含有反应产物的混合溶液;使用水和二氯甲烷的混合液对含有反应物的混合液ⅲ对含有反应产物的混合溶液进行萃取,使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅳ;使用溶剂ⅲ为淋洗剂,将粗反应产物ⅳ经柱层析纯化,得到反应产物ⅳ,即为含氮杂环化合物树枝取代的二溴苯中间体;
步骤五中所述的水和二氯甲烷的混合液中水和二氯甲烷的体积比为5:1;
步骤五中所述的溶剂ⅲ为石油醚和二氯甲烷的混合液,石油醚和二氯甲烷的体积比为8:1;
步骤五中所述的含氮杂环化合物树枝取代的二溴苯中间体的结构式为
六、在-100℃下将2mmol含氮杂环化合物树枝取代的二溴苯中间体和20ml四氢呋喃混合,再在搅拌条件下滴加1mmol正丁基锂,再在温度为-100℃下搅拌反应10h,再在搅拌的条件下滴加1mmol二苯基氯化膦,再在搅拌速度为60r/min下磁力搅拌12h,再使用水来淬灭反应,再使用水猝灭反应,再加入5mmol质量分数为30%的双氧水,再在搅拌速度为60r/min下搅拌1h,得到含有反应物的混合液ⅴ;使用二氯甲烷对含有反应物的混合液ⅴ进行萃取,然后使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物;以溶剂ⅴ为淋洗剂,将粗反应产物经柱层析纯化,再使用乙酸乙酯进行重结晶,得到含咔唑树枝蓝光热激发延迟荧光芳香材料;
步骤六中所述的溶剂ⅴ为石油醚和甲醇的混合液,石油醚和甲醇的体积比为50:1。
实施例一步骤一中得到的咔唑保护的质谱m/z:321.08(100.0%),322.08(20.5%),323.07(4.5%),323.08(2.0%)elementalanalysis:c,71.00;h,4.70;n,4.36;o,9.96;s,9.98。
实施例一步骤二中得到的3,6-双碘保护咔唑的质谱m/z:572.87(100.0%),573.87(20.5%),574.87(4.5%),574.88(2.0%)elementalanalysis:c,39.81;h,2.29;i,44.28;n,2.44;o,5.58;s,5.59。
实施例一步骤三中得到的含氮杂环化合物保护咔唑的质谱m/z:651.1980(100.0%),652.2014(46.5%),653.2048(10.6%),653.1938(4.5%),654.1972(2.1%),654.2081(1.6%),652.1951(1.1%)elementalanalysis:c,79.24;h,4.48;n,6.45;o,4.91;s,4.92。
实施例一步骤四中得到的含氮杂环化合物树枝的质谱m/z:497.18(100.0%),498.19(38.9%),499.19(7.4%),498.12(1.1%)elementalanalysis:c,86.90;h,4.66;n,8.44。
实施例一步骤五中得到的含氮杂环化合物树枝取代的二溴苯中间体的质谱m/z:1226.21(100.0%),1227.21(84.4%),1224.21(51.4%),1228.21(48.6%),1225.2184(43.4%),1229.21(41.0%),1228.21(35.1%),1226.28(18.1%),1230.217(17.1%),1229.22(9.6%),1227.21(4.9%),1231.22(4.7%),1227.21(2.2%),1230.22(1.9%),1228.21(1.9%),1225.21(1.1%),1229.20(1.1%),1228.25(1.0%)elementalanalysis:c,76.35;h,3.78;br,13.02;n,6.85。
实施例一步骤六中得到的含咔唑树枝蓝光热激发延迟荧光芳香材料的结构式为:
实施例二:本实施例与实施例一的不同点是:步骤三中所述的含氮杂环化合物为3,6-二叔丁基咔唑;步骤三中所述的含氮杂环化合物保护咔唑的结构式为
实施例二步骤三中所述的含氮杂环化合物保护咔唑
实施例二步骤四中所述的含氮杂环化合物树枝
实施例二步骤五中所述的含氮杂环化合物树枝取代的二溴苯中间体
实施例二步骤六中所述的含咔唑树枝蓝光热激发延迟荧光芳香材料
实施例三:本实施例与实施例一的不同点是:步骤五中将1mmol1,5-二溴-2,4-二氟苯、2.2mmol含氮杂环化合物树枝和2.27mmol碳酸钾加入到5ml二甲基亚砜中,再在氩气气氛和温度为130℃下反应6h,得到含有反应物的混合液ⅰ;使用水和二氯甲烷的混合液对含有反应物的混合液ⅰ进行萃取,然后使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅰ;以溶剂ⅰ为淋洗剂,将粗反应产物ⅰ经柱层析纯化,得到反应产物ⅰ,即为含氮杂环化合物树枝取代的二溴苯中间体;
步骤五中所述的溶剂ⅰ为石油醚和二氯甲烷的混合溶液,溶剂ⅰ中石油醚与二氯甲烷的体积比为5:1;
步骤五中所述的水和二氯甲烷的混合液中水和二氯甲烷的体积比为1:5;
步骤五中所述的含氮杂环化合物树枝取代的二溴苯中间体的结构式为
六、在-100℃下将2mmol含氮杂环化合物树枝取代的二溴苯中间体和20ml四氢呋喃混合,再在搅拌条件下滴加1mmol正丁基锂,再在温度为-100℃~0℃下搅拌反应10h~36h,再在搅拌的条件下滴加1mmol二苯基氯化膦,再在搅拌速度为60r/min下磁力搅拌10h,再使用水来淬灭反应,再使用水猝灭反应,再加入5mmol质量分数为30%的双氧水,再在搅拌速度为60r/min下搅拌1h,得到含有反应物的混合液ⅱ;使用二氯甲烷对含有反应物的混合液ⅱ进行萃取,然后使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅱ;以溶剂ⅱ为淋洗剂,将粗反应产物ⅱ经柱层析纯化,再使用乙酸乙酯进行重结晶,得到含咔唑树枝蓝光热激发延迟荧光芳香材料;
步骤六中所述的溶剂ⅱ为石油醚和甲醇的混合物,溶剂ⅱ中石油醚合甲醇的体积比为20:1。其他步骤及参数与实施例一均相同。
实施例三步骤五中得到的含氮杂环化合物树枝取代的二溴苯中间体的质谱m/z:1226.21(100.0%),1227.21(84.4%),1224.21(51.4%),1228.21(48.6%),1225.21(43.4%),1229.21(41.0%),1228.27(35.1%),1226.22(18.1%),1230.21(17.1%),1229.22(9.6%),1227.22(4.9%),1231.22(4.7%),1227.21(2.2%),1230.22(1.9%),1228.21(1.9%),1225.21(1.1%),1229.20(1.1%),1228.22(1.0%)elementalanalysis:c,76.35;h,3.78;br,13.02;n,6.85。
实施例六步骤六得到的含咔唑树枝蓝光热激发延迟荧光芳香材料的结构式为
实施例四:本实施例与实施例三的不同点是:步骤三中所述的含氮杂环化合物为3,6-二叔丁基咔唑;步骤三中所述的含氮杂环化合物保护咔唑的结构式为
实施例四步骤五中所述的含氮杂环化合物树枝取代的二溴苯中间体的质谱m/z:1675.71(100.0%),1674.71(84.1%),1676.72(58.9%),1673.71(51.4%),1677.71(48.6%),1672.71(43.2%),1676.71(40.9%),1674.72(30.3%),1678.71(28.7%),1677.72(23.0%),1675.72(11.8%),1679.72(11.2%),1678.72(6.6%),1676.72(3.4%),1680.72(3.2%),1676.71(2.2%),1675.71(1.9%),1679.73(1.5%),1677.71(1.3%),1676.72(1.3%),1674.71(1.1%),1678.71(1.1%),1675.72(1.1%)elementalanalysis:c,78.83;h,6.62;br,9.54;n,5.01。
实施例四步骤六中所述的含咔唑树枝蓝光热激发延迟荧光芳香材料的质谱m/z:1917.97(100.0%),1918.98(71.9%),1916.97(69.0%),1919.98(34.2%),1920.98(12.1%),1921.98(3.4%),1918.97(2.2%),1919.97(1.6%),1917.97(1.5%),1918.98(1.5%),1919.98(1.1%),1917.97(1.0%)elementalanalysis:c,83.89;h,6.83;n,4.38;o,1.67;p,3.23。
实施例五:本实施例与实施例一的不同点是:步骤五中将1mmol1,2-二溴-4,5-二氟苯、2.2mmol含氮杂环化合物树枝和2.27mmol碳酸钾加入到5ml二甲基亚砜中,再在氩气气氛和温度为130℃下反应6h,得到含有反应物的混合液ⅰ;使用水和二氯甲烷的混合溶液对含有反应物的混合液ⅰ进行萃取,然后使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅰ;以溶剂ⅰ为淋洗剂,将粗反应产物ⅰ经柱层析纯化,得到反应产物ⅰ;
步骤五中所述的溶剂ⅰ为石油醚和二氯甲烷的混合液,溶剂ⅰ中石油醚和二氯甲烷的体积比为5:1;
步骤五所述的水和二氯甲烷的混合溶液中水和二氯甲烷的体积比为1:5;
六、在-100℃下将2mmol反应产物ⅰ、20ml四氢呋喃和20ml乙醚混合,再在搅拌条件下滴加1mmol正丁基锂,再在温度为-100℃下搅拌反应10h,再在搅拌的条件下滴加1mmol二苯基氯化膦,再在搅拌速度为60r/min下磁力搅拌12h,再使用水来淬灭反应,得到含有反应物的混合液ⅱ;使用二氯甲烷对含有反应物的混合液ⅱ进行萃取,使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅱ;以溶剂ⅱ为淋洗剂,将粗反应产物ⅱ经柱层析纯化,再使用乙酸乙酯进行重结晶,得到反应产物ⅱ;
步骤六中所述的四氢呋喃与乙醚的体积比为1:1;
步骤六中所述的溶剂ⅱ为石油醚和二氯甲烷的混合溶液,溶剂ⅱ中石油醚和二氯甲烷的体积比为5:1;
七、在-100℃~0℃下将2mmol反应产物ⅱ和20ml四氢呋喃混合,再在搅拌的条件下滴加1mmol正丁基锂,再在温度为-100℃下搅拌反应12h,再在搅拌的条件下滴加1mmol二苯基氯化膦,再在搅拌速度为60r/min下磁力搅拌12h,再使用水来淬灭反应,再加入5mmol质量分数为30%的双氧水,再在搅拌速度为60r/min下搅拌1h,得到含有反应物的混合液ⅲ;使用二氯甲烷对含有反应物的混合液ⅲ进行萃取,然后使用无水硫酸钠对萃取后得到的有机层进行干燥,再使用旋转蒸发仪减压蒸发出溶剂,得到粗反应产物ⅲ;以溶剂ⅲ为淋洗剂,将粗反应产物ⅲ经柱层析纯化,再使用乙酸乙酯进行重结晶,得到含咔唑树枝蓝光热激发延迟荧光芳香材料;
步骤七中所述的溶剂ⅲ为石油醚和二氯甲烷的混合溶液,溶剂ⅲ中石油醚和二氯甲烷的体积比为5:1。
实施例五步骤五中所述的反应产物ⅰ的结构式为
实施例五步骤六中所述的反应产物ⅱ的结构式为
实施例五步骤七中得到的含咔唑树枝蓝光热激发延迟荧光芳香材料的结构式为
实施例六:本实施例与实施例五的不同点是:步骤三中所述的含氮杂环化合物为3,6-二叔丁基咔唑;步骤三中所述的含氮杂环化合物保护咔唑的结构式为
实施例六步骤五中所述的反应产物ⅰ的质谱m/z:1675.71(100.0%),1674.71(84.1%),1676.72(58.9%),1673.71(51.4%),1677.71(48.6%),1672.71(43.2%),1676.71(40.9%),1674.72(30.3%),1678.71(28.7%),1677.72(23.0%),1675.72(11.8%),1679.72(11.2%),1678.72(6.6%),1676.72(3.4%),1680.72(3.2%),1676.71(2.2%),1675.71(1.9%),1679.73(1.5%),1677.71(1.3%),1676.72(1.3%),1674.71(1.1%),1678.71(1.1%),1675.72(1.1%)elementalanalysis:c,78.83;h,6.62;br,9.54;n,5.01。
实施例六步骤六中所述的反应产物ⅱ的质谱m/z:1779.85(100.0%),1781.85(97.3%),1778.84(75.8%),1780.84(73.7%),1780.85(65.4%),1782.85(63.7%),1781.85(28.3%),1783.85(27.5%),1782.86(9.1%),1784.86(8.9%),1783.86(2.3%),1785.86(2.3%),1780.84(2.2%),1782.84(2.2%),1779.84(1.7%),1781.84(1.6%),1781.85(1.5%),1783.85(1.4%),1780.85(1.4%),1782.85(1.3%),1779.85(1.0%),1781.85(1.0%)elementalanalysis:c,82.27;h,6.79;br,4.49;n,4.72;p,1.74。
实施例六步骤七中得到的含咔唑树枝蓝光热激发延迟荧光芳香材料的m/z:1917.98(100.0%),1918.98(73.0%),1916.97(68.3%),1919.98(35.9%),1920.99(12.5%),1921.99(3.7%),1918.97(2.2%),1917.97(1.5%),1920.98(1.1%),1919.99(1.1%)elementalanalysis:c,83.89;h,6.83;n,4.38;o,1.67;p,3.23。
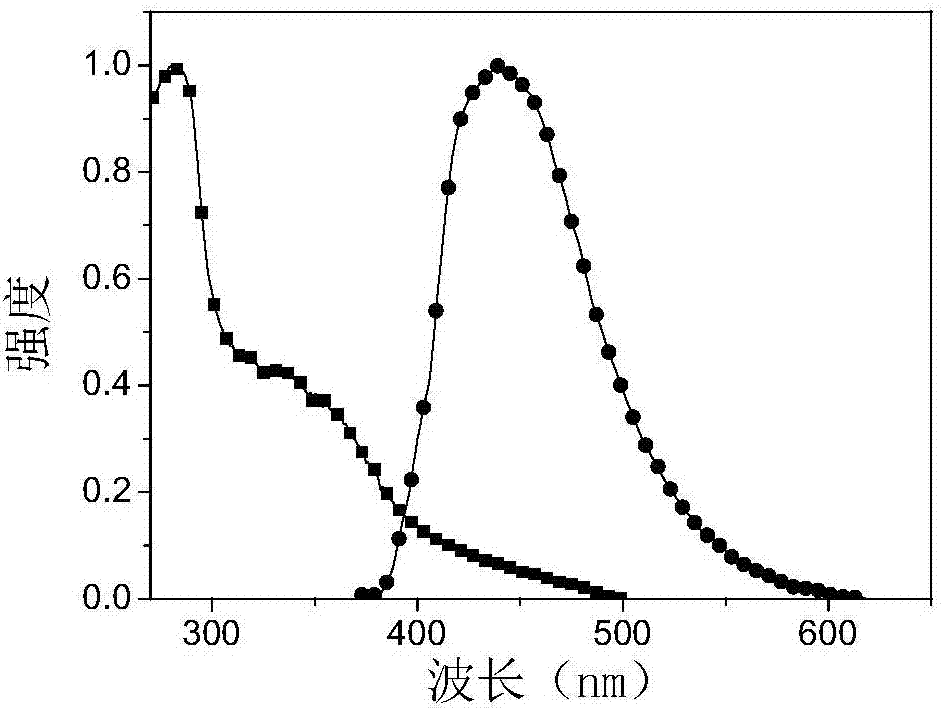
图1为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料溶于二氯甲烷中的光谱曲线,图1中“■”为紫外吸收光谱曲线,“▲”为荧光发射光谱曲线;
从图1可知,实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的紫外吸收光谱在375nm处,荧光发射光谱在460nm处。
图2为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的热重分析谱图;
取失重5%为裂解温度,从图2可知,实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的裂解温度为440℃。
图3为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料溶于二氯甲烷中的光谱曲线,图1中“■”为紫外吸收光谱曲线,“▲”为荧光发射光谱曲线;
从图3可知,实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的紫外吸收光谱在400nm处,荧光发射光谱在476nm处。
图4为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的热重分析谱图;
取失重5%为裂解温度,从图4可知,实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的裂解温度为445℃.
图5为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料溶于二氯甲烷中的光谱曲线,图1中“■”为紫外吸收光谱曲线,“▲”为荧光发射光谱曲线;
从图5可知,实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的紫外吸收光谱在404nm处,荧光发射光谱在464nm处。
图6为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的热重分析谱图;
取失重5%为裂解温度,从图6可知,实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的裂解温度为423℃。
图7为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料溶于二氯甲烷中的光谱曲线,图1中“■”为紫外吸收光谱曲线,“▲”为荧光发射光谱曲线;
从图7可知,实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的紫外吸收光谱在400nm处,荧光发射光谱在470nm处。
图8为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的热重分析谱图;
取失重5%为裂解温度,从图8可知,实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的裂解温度为423℃。
图9为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料溶于二氯甲烷中的光谱曲线,图1中“■”为紫外吸收光谱曲线,“▲”为荧光发射光谱曲线;
从图9可知,实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的吸收光谱在407nm处,荧光发射光谱在468nm处。
图10为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的热重分析谱图;
取失重5%为裂解温度,从图10可知,实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的裂解温度为413℃。
图11为实施例六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料溶于二氯甲烷中的光谱曲线,图1中“■”为紫外吸收光谱曲线,“▲”为荧光发射光谱曲线;
从图11可知,实施例六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料紫外吸收光谱在431nm处,荧光发射光谱在473nm处。
图12为实施例六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的热重分析谱图;
取失重5%为裂解温度,从图12可知,实施例六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的裂解温度为433℃。
应用试验一:含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中,热激发延迟荧光电致发光器件具体是按以下方法制备的:
一、将依次使用丙酮、无水乙醇和去离子水各清洗3次的ito基片安装到旋转蒸发仪上,再旋涂上pedot:pss,再在温度为120℃下烘烤10min,得到表面含有厚度为50nmpedot:pss薄膜的ito基片;
二、在表面含有厚度为50nmpedot:pss薄膜的ito基片上旋涂发光材料溶液,再在温度为100℃下烘烤10min,得到厚度为70nm的发光层;
步骤二中所述的涂发光材料溶液由发光材料和氯苯混合而成;所述的涂发光材料溶液中发光材料的质量与氯苯的体积比为10mg:1ml;所述的发光材料为dpetpo和实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料组成,且dpetpo与实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的摩尔比为3:1;
三、在真空度为1×10-6mbar和蒸镀速率为0.2nms-1的条件下在发光层上蒸镀tmpypb材料,在发光层上得到厚度为40nm的电子传输层;
四、在真空度为1×10-6mbar和蒸镀速率为0.2nms-1的条件下在电子传输层上蒸镀lif材料,在电子传输层上得到厚度为0.5nm的电子注入层;
五、在真空度为1×10-6mbar和蒸镀速率为0.2nms-1的条件下在电子注入层上蒸镀金属al材料,在电子注入层上得到厚度为150nm的阴极导电层,再进行封装,得到热激发延迟荧光电致发光器件。
应用试验二:本试验与应用试验一的不同点是:所述的发光材料为dpetpo和实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料组成,且dpetpo与实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的摩尔比为3:1。其他步骤与参数与应用试验一相同。
应用试验三:本试验与应用试验一的不同点是:所述的发光材料为dpetpo和实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料组成,且dpetpo与实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的摩尔比为3:1。其他步骤与参数与应用试验一相同。
应用试验四:本试验与应用试验一的不同点是:所述的发光材料为dpetpo和实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料组成,且dpetpo与实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的摩尔比为3:1。其他步骤与参数与应用试验一相同。
应用试验五:本试验与应用试验一的不同点是:所述的发光材料为dpetpo和实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料组成,且dpetpo与实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的摩尔比为3:1。其他步骤与参数与应用试验一相同。
应用试验六:本试验与应用试验一的不同点是:所述的发光材料为dpetpo和实施例六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料组成,且dpetpo与实施例六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料的摩尔比为3:1。其他步骤与参数与应用试验一相同。
图13为含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的电压-电流密度关系曲线,图13中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
从图13可知,应用试验一至应用试验六制备的热激发延迟荧光电致发光器件的电流密度分别为:72ma/cm2、78ma/cm2、82ma/cm2、93ma/cm2、75ma/cm2和62ma/cm2。
图14为含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的电压-亮度关系曲线,图14中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
从图14可知,应用试验一至应用试验六制备的热激发延迟荧光电致发光器件的亮度分别为2230cd/m2、1845cd/m2、3795cd/m2、5572cd/m2、1725cd/m2和1060cd/m2。
图15为含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的亮度-电流效率关系曲线,图15中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
从图15可知,应用试验一至应用试验六制备的热激发延迟荧光电致发光器件的电流效率分别为3.5ma/cm2、7.6ma/cm2、6.9ma/cm2、18.7ma/cm2、2.7ma/cm2和10.6ma/cm2。
图16为含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的亮度-功率效率关系曲线,图16中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
从图16可知,应用试验一至应用试验六制备的热激发延迟荧光电致发光器件的功率效率分别为3.5lm/w、5.2lm/w、6.5lm/w、17.0lm/w、4.2lm/w和9.7lm/w。
图17为含咔唑树枝蓝光热激发延迟荧光芳香材料作为客体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的亮度-外量子效率关系曲线,图17中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
从图17可知,应用试验一至应用试验六制备的热激发延迟荧光电致发光器件的外量子效率分别为3.0%、5.4%、5.4%、12.2%、3.2%和8.2%。
应用试验七:含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在热激发延迟荧光电致发光器件中,热激发延迟荧光电致发光器件具体是按以下方法制备的:
一、将依次使用丙酮、无水乙醇和去离子水各清洗3次的ito基片安装到旋转蒸发仪上,再旋涂上pedot:pss,再在温度为120℃下烘烤10min,得到表面含有厚度为50nmpedot:pss薄膜的ito基片;
二、在表面含有厚度为50nmpedot:pss薄膜的ito基片上旋涂发光材料溶液,再在温度为100℃下烘烤10min,得到厚度为20nm的发光层;
步骤二中所述的涂发光材料溶液由发光材料和氯苯混合而成;所述的涂发光材料溶液中发光材料的质量与氯苯的体积比为10mg:1ml;所述的发光材料为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和dmac-dps组成,且实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与dmac-dps的摩尔比为8:1;
三、在真空度为1×10-6mbar和蒸镀速率为0.2nms-1的条件下在发光层上蒸镀tmpypb材料,在发光层上得到厚度为40nm的电子传输层;
四、在真空度为1×10-6mbar和蒸镀速率为0.2nms-1的条件下在电子传输层上蒸镀lif材料,在电子传输层上得到厚度为0.5nm的电子注入层;
五、在真空度为1×10-6mbar和蒸镀速率为0.2nms-1的条件下在电子注入层上蒸镀金属al材料,在电子注入层上得到厚度为150nm的阴极导电层,再进行封装,得到热激发延迟荧光电致发光器件。
应用试验八:本试验与应用试验七的区别点是:所述的发光材料为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和dmac-dps组成,且实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与dmac-dps的摩尔比为8:1。其他步骤与参数与应用试验七相同。
应用试验九:本试验与应用试验七的区别点是:所述的发光材料为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和dmac-dps组成,且实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与dmac-dps的摩尔比为8:1。其他步骤与参数与应用试验七相同。
应用试验十:本试验与应用试验七的区别点是:所述的发光材料为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和dmac-dps组成,且实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与dmac-dps的摩尔比为8:1。其他步骤与参数与应用试验七相同。
应用试验十一:本试验与应用试验七的区别点是:所述的发光材料为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和dmac-dps组成,且实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与dmac-dps的摩尔比为8:1。其他步骤与参数与应用试验七相同。
应用试验十二:本试验与应用试验七的区别点是:所述的发光材料为实施例六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和dmac-dps组成,且实施例六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与dmac-dps的摩尔比为8:1。其他步骤与参数与应用试验七相同。
对应用试验七、应用试验八、应用试验九、应用试验十、应用试验十一和应用试验十二制备的热激发延迟荧光电致发光器件的亮度-外量子效率关系进行测试,如图19所示;
图19为含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的亮度-外量子效率关系曲线,图19中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
从图19可知,应用试验七至应用试验十二制备的热激发延迟荧光电致发光器件的亮度-外量子效率分别为11.4%、13.8%、11.5%、10.5%、10.2%和11.7%。
对应用试验七、应用试验八、应用试验九、应用试验十、应用试验十一和应用试验十二制备的热激发延迟荧光电致发光器件的电致发光光谱进行测试,如图20所示;
图20为含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在热激发延迟荧光电致发光器件中电致发光器件的电致发光光谱,图20中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
从图20可知,应用试验七至应用试验十二制备的热激发延迟荧光电致发光器件的光谱分别为476nm、472nm、488nm、480nm、480nm和476nm。
应用试验十三:含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在磷光器件中,磷光器件具体是按以下方法制备的:
一、将依次使用丙酮、无水乙醇和去离子水各清洗3次的ito基片安装到旋转蒸发仪上,再旋涂上pedot:pss,再在温度为120℃下烘烤10min,得到表面含有厚度为50nmpedot:pss薄膜的ito基片;
二、在表面含有厚度为50nmpedot:pss薄膜的ito基片上旋涂发光材料溶液,再在温度为100℃下烘烤10min,得到厚度为20nm的发光层;
步骤二中所述的涂发光材料溶液由发光材料和氯苯混合而成;所述的涂发光材料溶液中发光材料的质量与氯苯的体积比为10mg:1ml;所述的发光材料为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为10:1;
三、在真空度为1×10-6mbar和蒸镀速率为0.2nms-1的条件下在发光层上蒸镀tmpypb材料,在发光层上得到厚度为40nm的电子传输层;
四、在真空度为1×10-6mbar和蒸镀速率为0.2nms-1的条件下在电子传输层上蒸镀lif材料,在电子传输层上得到厚度为0.5nm的电子注入层;
五、在真空度为1×10-6mbar和蒸镀速率为0.2nms-1的条件下在电子注入层上蒸镀金属al材料,在电子注入层上得到厚度为150nm的阴极导电层,再进行封装,得到热激发延迟荧光电致发光器件。
应用试验十四:本试验与应用试验十三的区别点是:所述的发光材料为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为10:1。其他步骤与参数与应用试验十三相同。
应用试验十五:本试验与应用试验十三的区别点是:所述的发光材料为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为10:1。其他步骤与参数与应用试验十三相同。
应用试验十六:本试验与应用试验十三的区别点是:所述的发光材料为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为10:1。其他步骤与参数与应用试验十三相同。
应用试验十七:本试验与应用试验十三的区别点是:所述的发光材料为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为10:1。
应用试验十八:本试验与应用试验十三的区别点是:所述的发光材料为实施例六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且实施例六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为10:1。其他步骤与参数与应用试验十三相同。
对应用试验十三、应用试验十四、应用试验十五、应用试验十六、应用试验十七和应用试验十八制备的磷光器件的亮度-外量子效率关系进行测试,如图21所示;
图21为含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在磷光器件中的亮度-外量子效率关系曲线,图21中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
从图21可知,应用试验十三、应用试验十四、应用试验十五、应用试验十六、应用试验十七和应用试验十八制备的磷光器件的外量子效率分别为14.6%、13.8%、11.5%、19.0%、16.7%和7.0%。
图22为含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在磷光器件中的电致发光光谱,图22中“■”为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“●”为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▲”为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“▼”为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,“◆”为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料,
从图22可知,应用试验十三、应用试验十四、应用试验十五、应用试验十六、应用试验十七和应用试验十八制备的磷光器件的光谱均在565nm。
应用试验十九:含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在暖白光器件中,暖白光器件的制备方法具体是按以下方法制备的:
一、将依次使用丙酮、无水乙醇和去离子水各清洗3次的ito基片安装到旋转蒸发仪上,再旋涂上pedot:pss,再在温度为120℃下烘烤10min,得到表面含有厚度为50nmpedot:pss薄膜的ito基片;
二、在表面含有厚度为50nmpedot:pss薄膜的ito基片上旋涂发光材料溶液,再在温度为100℃下烘烤10min,得到厚度为20nm的发光层;
步骤二中所述的涂发光材料溶液由发光材料和氯苯混合而成;所述的涂发光材料溶液中发光材料的质量与氯苯的体积比为10mg:1ml;所述的发光材料为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且实施一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为30:1;
三、在真空度为1×10-6mbar和蒸镀速率为0.2nms-1的条件下在发光层上蒸镀tmpypb材料,在发光层上得到厚度为40nm的电子传输层;
四、在真空度为1×10-6mbar和蒸镀速率为0.2nms-1的条件下在电子传输层上蒸镀lif材料,在电子传输层上得到厚度为0.5nm的电子注入层;
五、在真空度为1×10-6mbar和蒸镀速率为0.2nms-1的条件下在电子注入层上蒸镀金属al材料,在电子注入层上得到厚度为150nm的阴极导电层,再进行封装,得到热激发延迟荧光电致发光器件。
应用试验二十:本试验与应用试验十九的区别点为:所述的发光材料为实施例二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且实施二制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为30:1。其他步骤及参数与应用试验十九相同。
应用试验二十一:本试验与应用试验十九的区别点为:所述的发光材料为实施例三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且实施三制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为30:1。其他步骤及参数与应用试验十九相同。
应用试验二十二:本试验与应用试验十九的区别点为:所述的发光材料为实施例四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且实施四制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为30:1。其他步骤及参数与应用试验十九相同。
应用试验二十三:本试验与应用试验十九的区别点为:所述的发光材料为实施例五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且实施五制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为30:1。其他步骤及参数与应用试验十九相同。
应用试验二十四:本试验与应用试验十九的区别点为:所述的发光材料为实施例六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料和po-01组成,且实施六制备的含咔唑树枝蓝光热激发延迟荧光芳香材料与po-01的摩尔比为30:1。其他步骤及参数与应用试验十九相同。
对应用试验十九制备的暖白光器件的亮度-外量子效率关系进行测试,如图23所示;
图23为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在暖白光器件中的亮度-外量子效率关系曲线;
从图23可知,实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在暖白光器件白光器件的外量子效率为15.3%。
对应用试验十九制备的暖白光器件的电致发光光谱进行测试,如图24所示;
图24为实施例一制备的含咔唑树枝蓝光热激发延迟荧光芳香材料作为主体发光材料应用在暖白光器件中的电致发光光谱。
从图24可知,白光器件的光谱为488nm和556nm。
- 还没有人留言评论。精彩留言会获得点赞!