1,3‑二羟基丙酮的制备方法与流程
本发明涉及有机合成
技术领域:
,具体涉及1,3-二羟基丙酮的制备方法。
背景技术:
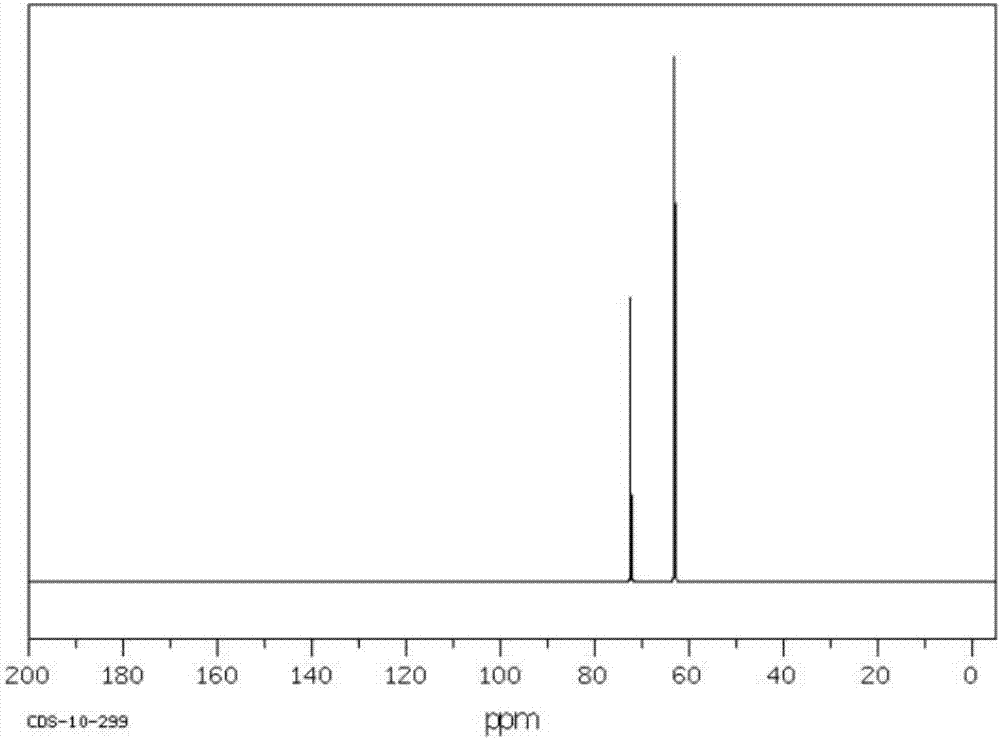
:1,3-二羟基丙酮(dihydroxyacetone,简称dha)是最简单的酮糖,它易溶于水、乙醇、丙酮和乙醚等有机溶剂。dha是一种重要的化工原料,广泛应用于精细化工、食品、化妆品和水质净化等行业;同时,dha也重要的有机合成中间体,可用于羟醛缩合制取多种手性化合物、还原制备具有光学活性的仲醇、经diels-alder加成制备糖类化合物或经进一步反应用于该类加成反应、与卡宾发生插入反应、与2,2-二烷氧基环丙烷衍生物作用制备内酯等。目前,1,3-二羟基丙酮主要通过微生物发酵法和甘油直接催化氧化法制备。微生物发酵法是使用醋酸杆菌将甘油发酵转化生成dha的方法。由于菌种对生存条件要求苛刻,其在储运过程中易丧失活性;而底物和产物在菌体细胞周围的高浓度积累则会延长发酵周期(300-400小时)并降低产率。因此,微生物发酵法的成本较高。甘油直接催化氧化法是在催化剂和氧化剂的共同作用下,通过一步反应将甘油转化为二羟基丙酮的方法。hiroshikimura等在以pt/c、pt-bi/c和pt-bi-ce/c为催化剂,最高可使70%的甘油发生转化,但dha的选择性过低(appl.catal.,a,1993,96,217-228.)。cn102432445a则公开了一种利用双氧水选择性氧化甘油制备dha的方法,但该方法中dha的选择性仍较低,仅达到24.2~85.7%。综合以上分析,现有的dha制备工艺仍有改进空间,需进一步提高甘油的有效利用率、降低dha的生产成本。技术实现要素:本发明的目的是针对现有技术的不足,提供一种1,3-二羟基丙酮的制备方法,该制备方法的合成周期短、效率高、成本低。为了实现上述目的,本发明提供一种1,3-二羟基丙酮的制备方法,包括以下步骤:(1)在催化剂存在的条件下,将甘油和卤代试剂进行接触反应,制备得到1,3-二氯-2-丙醇;(2)将1,3-二氯-2-丙醇进行氧化脱氢反应,得到中间产物1,3-二氯-2丙酮;(3)将1,3-二氯-2丙酮与碱性物质在含水介质中接触进行水解反应,得到1,3-二羟基丙酮,其中,所述水解反应的温度为25~60℃。通过上述技术方案,本发明提供了一种1,3-二羟基丙酮的制备方法,采用甘油为原料,甘油经催化卤代、氧化脱氢和水解反应制备得到1,3-二羟基丙酮。本发明中甘油的转化率以及1,3-二羟基丙酮的产率都较高,且此法采用氧化锆作催化剂,效率高、成本低,具有工业应用前景。本发明的其它特征和优点将在随后的具体实施方式部分予以详细说明。附图说明附图是用来提供对本发明的进一步理解,并且构成说明书的一部分,与下面的具体实施方式一起用于解释本发明,但并不构成对本发明的限制。在附图中:图1是本发明中甘油的质谱图;图2是本发明中甘油的核磁共振13c谱;图3是实施例1中1,3-二氯-2-丙醇的核磁共振13c谱;图4是实施例1中1,3-二氯-2-丙醇的质谱图;图5是实施例1中1,3-二氯丙酮的核磁共振13c谱;图6是实施例1中1,3-二氯丙酮的质谱图;图7是实施例1中1,3-二羟基丙酮的核磁共振13c谱。具体实施方式以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。本发明提供了一种1,3-二羟基丙酮的制备方法,包括以下步骤:(1)在催化剂存在的条件下,将甘油和卤代试剂进行接触反应,制备得到1,3-二氯-2-丙醇;(2)将1,3-二氯-2-丙醇进行氧化脱氢反应,得到中间产物1,3-二氯-2丙酮;(3)将1,3-二氯-2丙酮与碱性物质在含水介质中接触进行水解反应,得到1,3-二羟基丙酮,其中,所述水解反应的温度为25~60℃。根据本发明,具体的,在步骤(1)中,所述接触反应为:将甘油和催化剂在有机溶剂中按比例混合均匀,形成混合体系,再向混合体系中滴加卤代试剂,然后在50~120℃下,反应1~10h。根据本发明,本发明中所采用的有机溶剂是能够和甘油互溶的有机溶剂,可以为醇、醚、酮和胺等中的至少一种,优选为c1~c5的醇、c2~c5的醚和酮中的至少一种,更具体的,可以为甲醇、乙醇、异丙醇、正丁醇、丙酮、n-甲基吡咯烷酮、乙醚、甲基叔丁基醚、乙烯醚、乙酰胺、二亚甲砜和邻甲酚中的至少一种。根据本发明,在步骤(1)中,所述卤代试剂采用滴加的方式与甘油缓慢混合,避免将卤代试剂一次性加入混合体系中,能够有效降低混合体系中卤代试剂的浓度,降低副产物的产率,提高卤代试剂的转化率,降低反应体系中游离卤素的含量,降低游离卤素对环境的危害,优选的,所述卤代试剂的滴加速率为6~12ml/min。根据本发明,在步骤(1)中,为了提高甘油在卤代反应中的转化率,且降低副产物的产量,应该合理的控制甘油和卤代烃的配比,优选为所述甘油和卤代试剂的的摩尔比为1.0:(1.0~3.0)。根据本发明,催化剂的含量是影响催化反应效率的重要因素之一,在步骤(1)中,为了提高催化反应效率,使催化剂的催化效果达到最佳,进一步优选的,甘油和催化剂的质量比为1:(0.01~0.8)。根据本发明,进一步的,在步骤(1)中,所述催化剂为负载硫酸的氧化锆催化剂,该催化剂是一种固体负载超强酸催化材料,具有催化活性高、选择型好、不污染环境、不腐蚀设备,且可以重复利用,成本相对较低的特点。根据本发明,进一步的,在步骤(1)中,所述卤代试剂为卤化氢、卤化磷和二氯亚砜中的至少一种,如氯化氢、溴化氢、三氯化磷、五氯化磷和二氯亚砜中的至少一种,为了使卤化试剂能够在有机溶剂中更好的与甘油混合,进一步的,所述卤化试剂为液态,如盐酸、氢溴酸、三氯化磷、五氯化磷水溶液、二氯亚砜的乙醇溶液。根据本发明,进一步的,在步骤(2)中,所述氧化脱氢的方法为将1,3-二氯-2-丙醇、溴化钠与二氯异氰尿酸钠溶解于有机溶剂中,得到混合溶液,向混合溶液中加入ph缓冲剂,调节溶液ph为8.0~9.5,在搅拌的条件下,向混合溶液中滴加次氯酸钠。根据本发明,在步骤(2)中,由于1,3-二氯-2-丙醇的氧化脱氢反应释放大量的热量,为了降低体系热量,进一步的,所述氧化脱氢反应在冰水浴中进行。根据本发明,进一步的,在步骤(2)中,所述氧化脱氢反应的温度0~28℃,时间为30~180min。根据本发明,1,3-二氯-2-丙醇的氧化脱氢反应中,体系ph值对反应的影响比较大。当体系为酸性时,会产生氯气,从而降低目的产物选择性。因此,通过加入碱性缓冲剂,调节溶液的ph值,进一步的,所述缓冲剂为碳酸氢钠和碳酸钠缓冲液、硼酸-硼砂缓冲液、甘氨酸-氢氧化钠缓冲液、硼砂-氢氧化钠缓冲液中的一种。根据本发明,本发明的产物1,3-二羟基丙酮的沸点为70~75℃。因此,在步骤(3)中,本发明可以采用蒸馏的方法提纯1,3-二羟基丙酮,进一步的,所述蒸馏的温度为70~80℃。以下将通过实施例对本发明进行详细描述。本发明中物质的核磁共振13c谱通过美国varian公司生产的unityinova500mhz核磁共振波谱仪(5mm双共振探头,φ4mmzro2转子)测试得到;其质谱图通过agilent5977a型gc-mas测试得到。本发明中甘油、溴化钠、二氯异氰尿酸钠、次氯酸钠、碳酸氢钠和碳酸钠、氢氧化钠、三氯化磷、五氯化磷和二氯亚砜均购自国药集团化学试剂有限公司。其中,甘油的核磁共振13c谱如图1所示,其高分辨质谱图如图2所示,13cnmr谱图中化学位移为63.0和72.4处的峰分别归属于端位碳和中间碳,质谱图中质荷比为61的峰为标准峰。本发明中甘油的转化率通过以下公式计算得到:甘油转化率(%)=(m1-m2)/m1×100%1,3-二羟基丙酮的产率(%)=m实际/m理论×100%其中:m1为甘油的加入量,m2为步骤(1)反应结束后,甘油的剩余量;m理论是1,3-二羟基丙酮的理论生成量,m实际是1,3-二羟基丙酮的实际生成量。实施例1将0.2mol甘油和1g催化剂在100ml丙酮中混合均匀,形成混合体系(所述催化剂为负载硫酸的氧化锆,硫酸的质量为氧化锆质量的8%),再以9ml/min的速率向混合体系中滴加40ml质量分数为35%的盐酸,然后在80℃下反应2h,反应结束后,得到混合产物;过滤混合产物,收集滤渣(滤渣的主要成分是催化剂),将滤液用100ml水萃取3次,取下层溶液,对下层溶液进行减压蒸馏得到25.8g(0.2mol)1,3-二氯-2-丙醇,其核磁共振13c谱如图3所示,其高分辨质谱图如图4所示,13cnmr谱图中化学位移为45.8和71处的峰分别归属于端位碳和中间碳,质谱图中质荷比为79的峰为标准峰。将100ml水中置于冰水浴中,然后向水中加入0.2mol1,3-二氯-2-丙醇、0.3mol溴化钠与0.4mol二氯异氰尿酸钠,充分混合后得到混合溶液,向混合溶液中加入碳酸氢钠和碳酸钠缓冲液,调节溶液ph为9.0,在搅拌的条件下,向混合溶液中滴加70ml5m的次氯酸钠溶液,反应30min,将产物洗涤干燥后得到23.24g(0.183mol)1,3-二氯-2丙酮,其核磁共振13c谱如图5所示,其高分辨质谱图如图6所示,13cnmr谱图中化学位移为46.3和195处的峰分别归属于端位碳和中间碳,质谱图中质荷比为77的峰为标准峰。将0.183mol1,3-二氯-2丙酮加入100ml6m的氢氧化钠溶液中,在不断搅拌的条件下,在50℃下水解1h,反应结束后,将体系转移至蒸馏烧瓶中,在78℃下蒸馏得到15.35g(0.171mol)1,3-二羟基丙酮,其质谱图如图7所示,图中质荷比为145的峰为标准峰。实施例2将0.2mol甘油和1.2g催化剂在100mln-甲基吡咯烷酮中混合均匀,形成混合体系(所述催化剂为负载硫酸的氧化锆,硫酸的质量为氧化锆质量的10%),再以6ml/min的速率向混合体系中滴加26ml质量分数为40%的氢溴酸,然后在60℃下反应5h,反应结束后,得到混合产物;过滤混合产物,收集滤渣,将滤液用100ml水萃取3次,取下层溶液,对下层溶液进行减压蒸馏得到23.64g(0.183mol)1,3-二氯-2-丙醇;将100ml水中置于冰水浴中,然后向水中加入0.18mol1,3-二氯-2-丙醇、0.18mol溴化钠与0.27mol二氯异氰尿酸钠,充分混合后得到混合溶液,向混合溶液中加入甘氨酸-氢氧化钠缓冲液,调节溶液ph为9.5,在搅拌的条件下,向混合溶液中滴加100ml5m的次氯酸钠溶液,反应30min,将产物洗涤干燥后得到20.1g(0.158mol)1,3-二氯-2丙酮;将0.158mol1,3-二氯-2丙酮加入100ml6m的氢氧化钠溶液中,在不断搅拌的条件下,在30℃下进行水解反应1.5h,反应结束后,将体系转移至蒸馏烧瓶中,在75℃下蒸馏得到13.1g(0.145mol)1,3-二羟基丙酮。实施例3将0.2mol甘油和0.5g催化剂在100ml甲苯中混合均匀,形成混合体系(所述催化剂为负载硫酸的氧化锆,硫酸的质量为氧化锆质量的3%),再以9ml/min的速率向混合体系中滴加45ml三氯化磷,然后在50℃下反应10h,反应结束后,得到混合产物;过滤混合产物,收集滤渣,将滤液用100ml水萃取3次,取下层溶液对下层溶液进行减压蒸馏得到24.4g(0.189mol)1,3-二氯-2-丙醇;将100ml水中置于水浴锅中,调节水浴锅温度为10℃,然后向水中加入0.18mol1,3-二氯-2-丙醇、0.36mol溴化钠与0.54mol二氯异氰尿酸钠,充分混合后得到混合溶液,向混合溶液中加入硼砂-氢氧化钠缓冲液,调节溶液ph为8.7,在搅拌的条件下,向混合溶液中滴加30ml6m的次氯酸钠溶液,反应30min,将产物洗涤干燥后得到17.27g(0.136mol)1,3-二氯-2丙酮;将0.136mol1,3-二氯-2丙酮加入100ml6m的碳酸钠溶液中,在不断搅拌的条件下,在25℃下进行水解15min,反应结束后,将体系转移至蒸馏烧瓶中,在78℃下蒸馏得到11.4g(0.127mol)1,3-二羟基丙酮。实施例4将0.2mol甘油和0.8g催化剂在100ml甲基叔丁基醚中混合均匀,形成混合体系(所述催化剂为负载硫酸的氧化锆,硫酸的质量为氧化锆质量的5%),再以10ml/min的速率向混合体系中滴加40ml质量分数为30.2%的盐酸,然后在80℃下反应3h,反应结束后,得到混合产物;过滤混合产物,收集滤渣,将滤液用100ml水萃取3次,取下层溶液,对下层溶液进行减压蒸馏得到22g(0.17mol)1,3-二氯-2-丙醇;将100ml水中置于水浴锅中,调节水浴锅温度为18℃,然后向水中加入0.17mol1,3-二氯-2-丙醇、0.36mol溴化钠与0.45mol二氯异氰尿酸钠,充分混合后得到混合溶液,向混合溶液中加入硼酸-硼砂缓冲液,调节溶液ph为8.5,在搅拌的条件下,向混合溶液中滴加50ml6m次氯酸钠,反应30min,将反应产物洗涤干燥后得到19.56g(0.154mol)1,3-二氯-2丙酮;将0.165mol1,3-二氯-2丙酮加入100ml6m的氢氧化钠溶液中,在不断搅拌的条件下,在40℃下进行水解1.5h,反应结束后,将体系转移至蒸馏烧瓶中,在80℃下蒸馏得到12.28g(0.136mol)1,3-二羟基丙酮。实施例5将0.2mol甘油和0.3g催化剂在200ml异丙醇和100ml正丁醇的混合溶剂中混合均匀,形成混合体系(所述催化剂为负载硫酸的氧化锆,硫酸的质量为氧化锆质量的15%),再以12ml/min的速率向混合体系中滴加65ml质量分数为60%的二氯甲砜苯乙醇溶液,然后在120℃下反应1h,反应结束后,得到混合产物;过滤混合产物,收集滤渣,将滤液用100ml水萃取3次,取下层溶液,对下层溶液进行减压蒸馏得到19.37g(0.15mol)1,3-二氯-2-丙醇;将100ml水中置于置于水浴锅中,调节水浴锅温度为28℃,然后向水中加入0.15mol1,3-二氯-2-丙醇、0.075mol溴化钠与0.15mol二氯异氰尿酸钠,充分混合后得到混合溶液,向混合溶液中加入为碳酸氢钠和碳酸钠缓冲液,调节溶液ph为9.0,在搅拌的条件下,向混合溶液中滴加30ml4m次氯酸钠溶液,反应30min,将产物洗涤干燥后得到17.65g(0.139mol)1,3-二氯-2丙酮;将0.139mol1,3-二氯-2丙酮加入100ml4m的氢氧化钠溶液中,在不断搅拌的条件下,在60℃下进行水解30min,反应结束后,将体系转移至蒸馏烧瓶中,在70℃下减压蒸馏得到9.77g(0.108mol)1,3-二羟基丙酮。实施例6按照实施例1的方法,不同的是,催化剂为氧化锆,且催化反应时间为12h,具体方法如下:将0.2mol甘油和1g氧化锆在100ml丙酮中混合均匀,形成混合体系,再以9ml/min的速率向混合体系中滴加40ml质量分数为35%的盐酸,然后在80℃下反应24h,反应结束后得到混合产物;过滤混合产物,收集滤渣,将滤液用100ml水萃取3次,取下层溶液,对下层溶液进行减压蒸馏得到15.2g(0.118mol)1,3-二氯-2-丙醇;将100ml水中置于冰水浴中,然后向水中加入0.118mol1,3-二氯-2-丙醇、0.045mol溴化钠与0.06mol二氯异氰尿酸钠,充分混合后得到混合溶液,向混合溶液中加入碳酸氢钠和碳酸钠缓冲液,调节溶液ph为9.0,在搅拌的条件下,向混合溶液中滴加10ml5m的次氯酸钠溶液,反应30min,将产物洗涤干燥后得到10.3g(0.08mol)1,3-二氯-2丙酮;将0.125mol1,3-二氯-2丙酮加入10ml6m的氢氧化钠溶液中,在不断搅拌的条件下,在50℃下进行水解1h,反应结束后,将体系转移至蒸馏烧瓶中,在78℃下蒸馏得到4.6g(0.05mol)1,3-二羟基丙酮。实施例7按照实施例1的方法,不同的是,不采用缓冲溶液调节溶液ph,具体方法如下:将0.2mol甘油和1g催化剂在100ml丙酮中混合均匀,形成混合体系(所述催化剂为负载硫酸的氧化锆,硫酸的质量为氧化锆质量的8%),再以9ml/min的速率向混合体系中滴加40ml质量分数为35%的盐酸,然后在80℃下反应2h,反应结束后,得到混合产物;过滤混合产物,收集滤渣,将滤液用100ml水萃取3次,取下层溶液,对下层溶液进行减压蒸馏得到25.2g(0.195mol)1,3-二氯-2-丙醇;将100ml水中置于冰水浴中,然后向水中加入0.195mol1,3-二氯-2-丙醇、0.3mol溴化钠与0.4mol二氯异氰尿酸钠,充分混合后得到混合溶液,在搅拌的条件下,向混合溶液中滴加70ml5m的次氯酸钠溶液,反应30min,将产物洗涤干燥后得到10.8g(0.085mol)1,3-二氯-2丙酮;将0.085mol1,3-二氯-2丙酮加入100ml6m的氢氧化钠溶液中,在不断搅拌的条件下,在50℃下进行水解1h,反应结束后,反应结束后,将体系转移至蒸馏烧瓶中,在78℃下蒸馏得到6.81g(0.076mol)1,3-二羟基丙酮。对比例1采用中国专利cn102432445a中的方法制备1,3-二羟基丙酮,具体方法如下:将2.42kgcu(no3)2·3h2o、5.82kgni(no3)2·6h2o和3.75kgal(no3)3·9h2o用去离子水配成40l溶液;将1.06kgna2co3用去离子水配成40l溶液,两溶液混合后,60℃下剧烈搅拌0.5小时,并加naoh控制混液ph值为10.0。所得的胶体过滤或离心脱水,水洗至中性,100℃烘干后,置于500℃下n2气氛中煅烧2小时,所得样品于质量百分比浓度为40%的甘油水溶液中,ph值控制在10.0,80℃下搅拌24h,所得胶体过滤或离心脱水,100℃烘干,得所需催化剂。取质量百分比浓度为40%的甘油水溶液5l,加入甘油质量20%的所制催化剂,升温至50℃后,开始滴加质量百分比浓度为3.0%的双氧水,反应24h后将催化剂和反应液离心分离,得到1,3-二氯-2丙酮。对比例2按照实施例1的方法,不同的是,步骤(3)中水解反应的温度为20℃,具体方法如下:将0.2mol甘油和1g催化剂在100ml丙酮中混合均匀,形成混合体系(所述催化剂为负载硫酸的氧化锆,硫酸的质量为氧化锆质量的8%),再以9ml/min的速率向混合体系中滴加40ml质量分数为35%的盐酸,然后在80℃下反应2h,反应结束后,得到混合产物;过滤混合产物,收集滤渣,将滤液用100ml水萃取3次,取下层溶液,对下层溶液进行减压蒸馏得到24.6g(0.191mol)1,3-二氯-2-丙醇;将100ml水中置于冰水浴中,然后向水中加入0.191mol1,3-二氯-2-丙醇、0.3mol溴化钠与0.4mol二氯异氰尿酸钠,充分混合后得到混合溶液,向混合溶液中加入碳酸氢钠和碳酸钠缓冲液,调节溶液ph为9.0,在搅拌的条件下,向混合溶液中滴加70ml5m的次氯酸钠溶液,反应30min,将产物洗涤干燥后得到23.49g(0.185mol)1,3-二氯-2丙酮;将0.185mol1,3-二氯-2丙酮加入100ml6m的氢氧化钠溶液中,在不断搅拌的条件下,在20℃下水解1h,反应结束后,将体系转移至蒸馏烧瓶中,在78℃下蒸馏得到1.62g(0.018mol)1,3-二羟基丙酮。对比例3将0.2mol甘油和1g催化剂在100ml丙酮中混合均匀,形成混合体系(所述催化剂为负载硫酸的氧化锆,硫酸的质量为氧化锆质量的8%),再以9ml/min的速率向混合体系中滴加40ml质量分数为35%的盐酸,然后在80℃下反应2h,反应结束后,得到混合产物;过滤混合产物,收集滤渣,将滤液用100ml水萃取3次,取下层溶液,对下层溶液进行减压蒸馏得到25.3g(0.196mol)1,3-二氯-2-丙醇;将100ml水中置于冰水浴中,然后向水中加入0.196mol1,3-二氯-2-丙醇、0.3mol溴化钠与0.4mol二氯异氰尿酸钠,充分混合后得到混合溶液,向混合溶液中加入碳酸氢钠和碳酸钠缓冲液,调节溶液ph为9.0,在搅拌的条件下,向混合溶液中滴加70ml5m的次氯酸钠溶液,反应30min,将产物洗涤干燥后得到23.9g(0.188mol)1,3-二氯-2丙酮;将0.188mol1,3-二氯-2丙酮加入100ml6m的氢氧化钠溶液中,在不断搅拌的条件下,在80℃下水解20min,反应结束后,将体系转移至蒸馏烧瓶中,在78℃下蒸馏得到3.15g(0.035mol)1,3-二羟基丙酮。通过实施例1-7以及对比例1-3中的方法制备1,3-二羟基丙酮,其中甘油转化率、1,3-二羟基丙酮产率以及整个反应周期的数据汇集与表1。表1实施例编号甘油转化率/%1,3-二羟基丙酮产率/%反应周期实施例110085.3约6h实施例291.672.9约10h实施例394.663.4约12h实施例485.368.8约6h实施例575.154.3约4h实施例658.925约28h实施例797.537.8约6h对比例1--55h对比例295.59约6h对比例39817.5约6h实验结构表明:本发明中甘油的转化率高,且最终产物1,3-二羟基丙酮产率也很高,达到54.3~85.3%,且本发明中其制备方法的周期短,采用价格较低的氧化锆作为催化剂,无需使用昂贵的催化剂,降低了生产成本,在1,3-二羟基丙酮的工业化生产上具有广阔的应用前景。以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合。为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1