构建待测基因组的DNA测序文库的方法及其应用与流程
本发明涉及生物技术领域。具体地,本发明涉及构建待测基因组的dna测序文库的方法及其应用。更具体地,本发明涉及构建待测基因组的dna测序文库的方法、确定待测基因组的dna序列信息的方法以及确定待测基因组三维空间结构的方法。
背景技术:
近年来随着基因组学的发展,人类基因组计划成功绘制了人类基因组dna序列图谱。人类基因组百科全书计划相关研究,分析发现了几万个基因,几十万个不同的基因调控元件。最近的研究表明,基因组的三维空间结构对基因组的表达、调控等功能有重要的影响。因此,如何获得不同细胞种类在不同细胞周期,在不同发育和分化阶段以及在正常细胞向疾病细胞转变的过程中基因组的三维结构信息成为了亟待解决的问题。而染色质构象捕获技术和二代测序技术的发展普及,使得基于二代测序的全基因组染色质构象捕获技术hi-c(wholegenomechromosomeconformationcapture)成为了研究染色体三维结构的重要手段。然而,全基因组染色质构象捕获技术极为受限于细胞数目,如果不能获得足够量的包含足够多有效连接事件的用于建库的dna,是无法得到有效以及足够高分辨率的基因组三维结构信息的。
从而,如何建立不易获得的、少量数量类型细胞的dna测序文库并有效用于dna测序和获得足够高分辨率的基因组三维结构信息,是有待解决的问题。
技术实现要素:
本申请是基于发明人对以下问题的发现而做出的:
传统的hi-c技术反应体系较大,离心、洗涤、换管步骤繁多,在dna扩增前进行片段大小选择会造成大量的dna损失。而且dna片段大小选择范围较窄,远远超过超声波打断对dna片段的富集程度,进一步减少了用于二代测序的dna量。以上的几点原因,造成了hi-c技术通常用于研究容易获得的数量级在107以上的常见类型细胞,对于处于胚胎发育早期的这种极难获得大量数目的细胞类型是无能为力的。基于发明人对以上传统技术问题的发现,发明人通过缩小反应体系、更改细胞转移方式、改变dna片段大小选择的范围和时间等,创造性地提出了改进的全基因组dna测序文库构建方法(在本申请中,简称为sishi-c(smallscaleinsituhi-c)技术),该方法可以用来研究细胞数目低至10个的珍贵细胞类型,为研究胚胎早期发育的基因组三维结构建立和基因表达调控带来了希望。
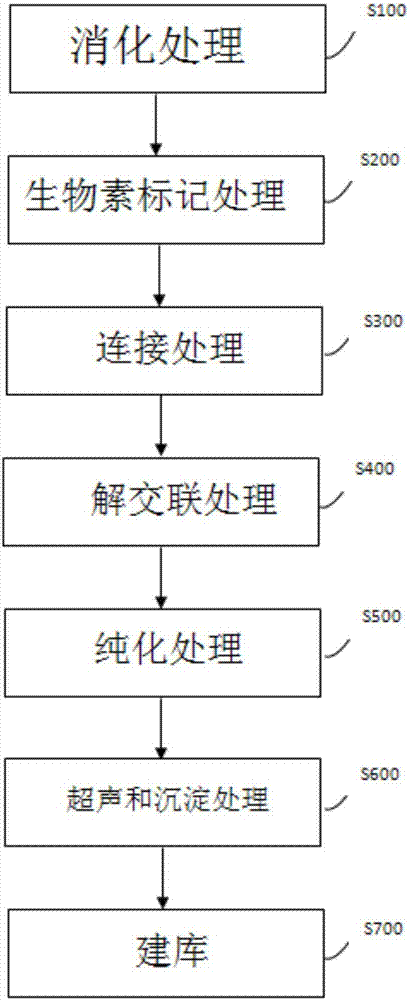
在本发明的第一方面,本发明提出了一种构建待测基因组的dna测序文库的方法。根据本发明的实施例,所述方法包括:(1)利用限制性内切酶对待测基因组进行消化处理,以便获得消化处理产物;(2)将所述消化处理产物进行生物素标记处理,以便获得生物素标记处理产物;(3)利用dna连接酶对所述生物素标记处理产物进行连接处理,以便获得连接产物;(4)将连接产物进行解交联处理;(5)将解交联处理产物进行纯化处理;(6)将纯化处理产物进行超声和沉淀处理,所述沉淀处理是通过将超声处理产物与链酶亲合素磁珠进行接触进行的,以便获得结合有链酶亲合素磁珠的目标dna片段;以及(7)基于结合有链酶亲合素磁珠的目标dna片段,进行建库。利用根据本发明实施例的构建待测基因组的dna测序文库的方法可以用来构建细胞数目低至10个细胞或基因组量低至50pg的dna测序文库,进而实现不易获得的、少量数量类型细胞的基因组染色质构象的捕获。
在本发明的第二方面,本发明提出了一种测序文库。根据本发明的实施例,所述测序文库是通过前面所述的构建待测基因组的dna测序文库的方法获得的。利用根据本发明实施例的测序文库进行测序,可获得细胞数目低至10个的细胞或基因组量低至50pg的基因组序列信息和足够高分辨率的基因组三维结构信息。
在本发明的第三方面,本发明提出了一种确定待测基因组的dna序列信息的方法。根据本发明的实施例,所述方法包括:根据前面所述的方法构建待测基因组的dna测序文库;对所述dna测序文库进行测序,以便获得测序结果;以及基于所述测序结果,确定所述待测基因组的dna序列信息。利用根据本发明实施例的确定待测基因组的dna序列信息的方法,可获得细胞数目低至10个的细胞或基因组量低至50pg的基因组序列信息。
在本发明的第四方面,本发明提出了一种用于确定待测基因组三维空间结构的方法。根据本发明的实施例,所述方法包括:根据前面所述的方法构建待测基因组的dna测序文库;对所述dna测序文库进行测序,以便获得测序结果;以及基于所述测序结果,确定所述待测基因组的三维空间结构信息。利用根据本发明实施例的确定待测基因组三维空间结构的方法,可获得细胞数目低至10个的细胞或基因组量低至50pg的足够高分辨率的基因组三维结构信息。
需要说明的是,本发明所提出的待测基因组是指细胞或组织的全基因组或部分基因组,并且基因组由染色质或染色体组成。本领域的技术人员可以理解,基因组的来源不受特别限制,可以从任何可能的途径获得,可以是通过市售直接获得,也可以是从其他实验室直接获取,还可以是直接从细胞或组织样本中提取的。
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
图1是根据本发明实施例的构建待测基因组的dna测序文库的方法的流程图;
图2是根据本发明又一实施例的构建待测基因组的dna测序文库的方法的流程图;
图3是根据本发明又一实施例的构建待测基因组的dna测序文库的方法的流程图;
图4是根据本发明实施例的truseq建库的流程图;
图5是根据本发明再一实施例的truseq建库的流程图;
图6是根据本发明再一实施例的truseq建库的流程图;
图7是根据本发明再一实施例的truseq建库的流程图;以及
图8是根据本发明实施例的验证本发明实施例所提出的建库方法具有显著优势的结果图。
具体实施方式
下面详细描述本发明的实施例,所述实施例的示例在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。
构建待测基因组的dna测序文库的方法
在本发明的第一方面,本发明提出了一种构建待测基因组的dna测序文库的方法。根据本发明的实施例,参考图1,该方法包括:s100:利用限制性内切酶对待测基因组进行消化处理,以便获得消化处理产物;s200:将所述消化处理产物进行生物素标记处理,以便获得生物素标记处理产物;s300:利用dna连接酶对所述生物素标记处理产物进行连接处理,以便获得连接产物;s400:将连接产物进行解交联处理;s500:将解交联处理产物进行纯化处理;s600:将纯化处理产物进行超声和沉淀处理,所述沉淀处理是通过将超声处理产物与链酶亲合素磁珠进行接触进行的,以便获得结合有链酶亲合素磁珠的目标dna片段;以及s700:基于结合有链酶亲合素磁珠的目标dna片段,进行建库。利用根据本发明实施例的构建待测基因组的dna测序文库的方法可以用来构建细胞数目低至10个的细胞或基因组量低至50pg的dna测序文库,进而实现不易获得的、少量数量类型细胞的基因组染色质构象的捕获。
根据本发明的实施例,参考图2,所述待测基因组是通过裂解细胞或组织而获得的,任选地,所述细胞为细胞系或原代细胞。裂解细胞或组织而释放细胞或组织中的基因组。
根据本发明的实施例,参考图3,所述细胞或组织预先经过甲醛交联处理。进而甲醛瞬时固定细胞或组织内自然状态下在空间上接近的dna-蛋白质和蛋白质-蛋白质复合物。
根据本发明的实施例,所述细胞或基因组由口吸管进行转移处理。发明人发现,使用口吸管精确转移细胞或组织可有效避免离心法更换溶液带来的细胞损失。
根据本发明的实施例,所述限制性内切酶为mboi。发明人发现,mboi的识别位点为gatc,碱基分布均匀,在基因组上进行切割时,不会有明显的偏好性;同时mboi的识别位点为四个碱基,相比于常用的六碱基限制性内切酶,其切割频率更高,理论上得到的数据的分辨率更高;最后mboi商业化程度高,使用成本低,可以轻松获得,因而有效控制了整个构建dna测序文库的成本。
根据本发明的实施例,所述生物素标记处理是通过如下方式进行的:将消化处理产物与三磷酸腺嘌呤脱氧核苷酸、三磷酸鸟嘌呤脱氧核苷酸、三磷酸胸腺嘧啶脱氧核甘酸、生物素标记的三磷酸胞嘧啶脱氧核苷酸以及dna聚合酶大片段进行接触,所述接触是在37℃的条件下进行1.5小时。通过上述方式可以将生物素高效标记到mboi酶切片段的末端。
根据本发明的实施例,所述连接处理为t4连接,所述解交联处理是通过将所述连接产物与蛋白酶k、sds以及氯化钠接触进行的。利用t4dna连接酶进行连接可将dna不同的切口末端连接成环状的嵌合分子,进而通过上述的解交联处理,将dna与其结合的蛋白质分开而将dna有效释放出来。
根据本发明的实施例,所述纯化处理是通过将所述解交联处理产物与预冷的无水乙醇进行接触进行的,所述接触是在-80℃的条件下进行15分钟。利用上述的纯化处理方式,dna有效沉淀于试管底部,进而实现dna的纯化。
根据本发明的实施例,所述纯化处理过程中将所述解交联处理产物与肝糖原和醋酸钠进行接触。发明人发现,在纯化dna的过程中,因为细胞数目少,dna量特别少,而加入肝糖原与dna共沉淀可以指示dna沉淀在ep管中的位置,进而有效防止了洗涤过程中dna损失。
根据本发明的实施例,所述超声是在peakpower为50,dutyfactor为20,cycles/burst为200的条件下进行134s。其中,peakpower表示最高入射功率,是作用在样品上的瞬时超声波功率;dutyfactor表示工作系数,即超声波作用于样品的时间占总时间段的百分数;cycles/burst表示超声波作用于样品过程中超声波能量传递的数目。发明人发现,在上述超声条件下,可有效减少dna片段大小选择过程中的dna损失。
根据本发明的实施例,参考图4,s700中的所述建库为truseq建库,s700包括将结合有链酶亲合素磁珠的目标dna片段进行s710:末端修复、s720:末端加三磷酸腺嘌呤脱氧核糖核苷酸和s730:连接测序接头序列处理。
根据本发明的实施例,s710末端修复处理之后、s720末端加三磷酸腺嘌呤脱氧核糖核苷酸处理之前进一步包括吐温洗涤所述磁珠处理;优选地,s720末端加三磷酸腺嘌呤脱氧核糖核苷酸处理之后、s730连接测序接头序列处理之前进一步包括吐温洗涤所述磁珠处理;优选地,s730连接测序接头序列处理之后进一步包括吐温洗涤所述磁珠处理。在每一步反应结束后,直接用吐温洗涤溶液洗涤磁珠两次,即可简单快速的更换溶液,避免了dna在纯化过程中的损失。
根据本发明的实施例,参考图5,进一步包括将连接测序接头序列处理产物进行s740:dna第一洗脱处理和s750:pcr扩增。经过第一洗脱处理,可将目标dna片段从链酶亲合素磁珠高效洗脱下来,经过pcr扩增,可实现目标dna的高效富集。
根据本发明的实施例,参考图6,进一步包括s760:将pcr扩增产物与ampurexp磁珠进行结合处理,任选地,参考图7,进一步包括s770:将结合有ampurexp磁珠的pcr扩增产物进行dna第二洗脱处理。从而得到大小在200碱基对到1000碱基对范围内的dna片段。
根据本发明实施例的上述方法获得的测序文库可作为sishi-c文库用于二代测序。
根据本发明实施例的上述方法使用很小的反应体系,在大大降低成本的同时增加了酶切和连接反应的效率;在细胞裂解前,使用口吸管精确转移细胞避免了离心法更换溶液带来的细胞损失;将dna结合在链霉亲合素磁珠上进行truseq建库、使用更温和的磁珠洗涤条件以及控制建库操作在同一个ep管中进行,这些措施成功绕开纯化回收步骤,并有效减少了在建库过程中的dna损失;优化超声打断dna片段的条件、两步法洗脱dna、在pcr之后将片段大小在200bp到1000bp范围内的dna全部保留下来,进一步提高了最后可利用的dna量。整个方法不但显著减少了起始细胞的用量,成本也仅为传统方法的十分之一,从而高效实现了基因组染色质构象捕获。
本领域技术人员可以理解的是,基于本申请所述的方法所获得的构建待测基因组的dna测序文库的设备、装置、单元、模块也在本申请的保护范围内,所述设备、装置、单元、模块的优点与前面所述的方法类似,在此不再详述。本领域技术人员能够理解的是,可以采用本领域中已知的任何适于进行上述操作的装置作为上述各个单元的组成部件。在本文中所使用的术语“相连”应作广义理解,可以是直接相连,也可以通过中间媒介间接相连,对于本领域的普通技术人员而言,可以根据具体情况理解上述术语的具体含义。
测序文库
在本发明的第二方面,本发明提出了一种测序文库。根据本发明的实施例,所述测序文库是通过前面所述的构建待测基因组的dna测序文库的方法获得的。利用根据本发明实施例的测序文库进行测序,可获得细胞数目低至10个的细胞或基因组量低至50pg的基因组序列信息和足够高分辨率的基因组三维结构信息。
确定待测基因组的dna序列信息的方法
在本发明的第三方面,本发明提出了一种确定待测基因组的dna序列信息的方法。根据本发明的实施例,该方法包括:根据前面所述的方法构建待测基因组的dna测序文库;对所述dna测序文库进行测序,以便获得测序结果;以及基于所述测序结果,确定所述待测基因组的dna序列信息。关于构建待测基因组的dna测序文库的方法,前面已经进行了详细描述,在此不再赘述。根据本发明的实施例,对测序文库进行测序的方法和装置不受特别限制,考虑到技术的成熟度,根据本发明的实施例,可以采用第二代测序技术,诸如solexa、solid和454测序技术。当然,也可以采用正在开发或者尚未开发的新型测序技术,例如单分子测序技术,诸如:helicos公司的truesinglemoleculednasequencing技术,pacificbiosciences公司的thesinglemolecule,real-time(smrt.tm.)技术,以及oxfordnanoporetechnologies公司的纳米孔测序技术等(rusk,nicole(2009-04-01).cheapthird-generationsequencing.naturemethods6(4):244–245)。
发明人惊奇地发现,利用根据本发明实施例的确定待测基因组的dna序列信息的方法,能够灵敏、准确、高效地确定微量细胞(细胞起始量低至10个)的基因组或微量基因组(dna起始量低至50pg)的基因组序列信息。
确定待测基因组三维空间结构的方法
在本发明的第四方面,本发明提出了一种用于确定待测基因组三维空间结构的方法。根据本发明的实施例,所述方法包括:根据前面所述的方法构建待测基因组的dna测序文库;对所述dna测序文库进行测序,以便获得测序结果;以及基于所述测序结果,确定所述待测基因组的三维空间结构信息。
根据本发明的具体实施例,所述方法包括根据前面所述的方法构建待测基因组的dna测序文库;对于所得到的文库进行双端测序,以得到文库中每个dna片段两端的序列信息;将两端的序列信息分别比对到基因组上,从而可以得到基因组中线性距离各异的每两个片段间的空间接近程度信息,进而结合数学方法推断出基因组的三维空间结构(aidenetal.comprehensivemappingoflong-rangeinteractionsrevealsfoldingprincipleofthehumangenome.science.2009)。
发明人惊奇地发现,利用根据本发明实施例的确定待测基因组染色质目标区域的序列信息的方法,能够灵敏、准确、高效地确定微量细胞(细胞起始量低至10个)基因组或微量基因组(dna起始量低至50pg)的足够高分辨率的基因组三维结构信息。
下面参考具体实施例,对本发明进行说明,需要说明的是,这些实施例仅是说明性的,而不能理解为对本发明的限制。
下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件(例如参考j.萨姆布鲁克等著,黄培堂等译的《分子克隆实验指南》,第三版,科学出版社)或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品,例如可以采购自illumina公司。
实施例1构建细胞基因组的dna测序文库
1.1试剂准备
裂解液(lysisbuffer)
·10mmtris-hcl.,ph=7.4
·10mmnacl
·0.5%np-40
·0.1mmedta
·1xproteinaseinhibitor
2x生物素和链霉亲合素磁珠结合溶液(2xbindingbuffer)
·10mmtris-hcl.,ph=8.0
·2mnacl
·1mmedta
吐温洗涤溶液(tweenwashbuffer)
·5mmtris-hcl.,ph=8.0
·1mnacl
·0.05%tween
·0.5mmedta
1.2甲醛交联
将收集的样品在体视镜下用口吸管转移到新鲜配制的含有1%甲醛的pbs溶液中,室温固定10分钟,加入2.5m甘氨酸溶液至终浓度为0.2m,室温静置10分钟。用口吸管将样品转移到pbs溶液中洗涤一次,然后转移到pcr管中。
1.3裂解样品及限制性内切酶消化
向含有样品的pcr管中加入50微升裂解液,并用低吸附带滤芯的枪头反复吹吸混匀,在冰上静置50分钟。离心去上清。加入10微升0.5%的sds溶液,62℃金属浴10分钟。
向样品中加入25微升水和10微升10%tritonx-100溶液。用移液器反复吹吸混匀,将样品置于金属浴中37℃孵育15分钟。混匀液体以防止后续sds对限制性内切酶活性的影响。
继续向样品中加入5微升10xnebbuffer2和50u限制性内切酶mboi,混匀后置于旋转混匀仪上,37℃酶切15小时,将染色质进行充分地消化和分离。
1.4末端标记和连接
第二天,将样品从旋转混匀仪上取下,置于62℃金属浴上静置20分钟,以失活mboi。在冰上,向每个样品中分别加入1mm三磷酸腺嘌呤脱氧核苷酸(datp),1mm三磷酸鸟嘌呤脱氧核苷酸(dgtp)和1mm三磷酸胸腺嘧啶脱氧核甘酸(dttp)各0.5微升,以及3.75微升生物素标记的0.4mm三磷酸胞嘧啶脱氧核苷酸(biotin-14-dctp)。混匀后加入10udna聚合酶大片段(klenow),将样品置于旋转混匀仪上37℃处理1.5小时,以将生物素标记到mboi酶切片段的末端。
将含有样品的溶液从pcr管中转移到低吸附的1.5毫升ep管中。并加入12微升10xnebt4dna连接酶反应溶液、7微升10%tritonx-100、1.2微升10毫克每微升的牛血清白蛋白、1微升400u/ult4dna连接酶和39微升水。混匀后,将样品置于恒温混匀仪(thermomixer)上,500转/分钟,24℃处理5.5小时,利用dna连接酶将不同的切口末端连接成环状的嵌合分子。
1.5解交联和dna纯化
连接反应结束后,向每个样品中加入100u蛋白酶k和12微升10%sds溶液,55℃金属浴30分钟。然后加入13微升5m氯化钠,在涡旋仪上混匀后置于65℃烘箱中,过夜解交联。
第三天,将样品从烘箱中取出,当温度降至室温后,向每个样品中加入1微升肝糖原和15微升3m醋酸钠,在涡旋仪上混匀,然后加入240微升预冷的无水乙醇,上下颠倒混匀,-80℃静置15分钟。高速离心,用75%无水乙醇洗涤沉淀两次,待沉淀干燥后用50微升水溶解纯化得到的dna,37℃金属浴15分钟。在纯化dna的过程中,因为细胞数目少,dna量特别少,加入肝糖原与dna共沉淀以指示dna沉淀在ep管中的位置,防止洗涤过程中dna损失。
1.6超声打断dna和生物素标记沉淀
用covarism220超声波dna破碎仪将样品dna剪切打碎成300-500碱基对(bp)大小的片段,超声条件为:peakpower50,dutyfactor20,cycles/burst200,time134s。用20微升水洗涤covaris超声管以减少dna损失。发明人发现,在上述超声条件下可以减少dna片段大小选择过程中的dna损失。
准备链霉亲和素磁珠(dynabeadsmyonestreptavidinc1),每个样品需要100微克磁珠。用吐温洗涤溶液清洗磁珠两次,然后用70微升2x生物素和链霉亲合素磁珠结合溶液重悬磁珠,并将其加入每个样品中,用移液器吹吸混匀。将样品置于旋转混匀仪上,室温50分钟,进行生物素标记沉淀以获取有标记的目的dna片段。
1.7建库和dna片段选择
经过生物素标记沉淀后,带有生物素标记的目标dna片段结合在链霉亲和素磁珠上。因为生物素标记的dna与链霉亲和素磁珠结合非常稳定,所以这些结合目标dna的磁珠可以直接用来进行truseq建库。依次进行末端修复、在dna片段末端加三磷酸腺嘌呤脱氧核糖核苷酸(datp)和连接测序接头序列。在每一步反应结束后,直接用吐温洗涤溶液洗涤磁珠两次,即可简单快速的更换溶液,避免了dna在纯化过程中的损失。
第四天,连接测序接头序列后,将样品置于磁力架上,待磁珠全部吸附到磁力架,溶液变澄清后,弃上清,用吐温洗涤溶液清洗磁珠两次,每次在旋转混匀仪上室温转动2分钟。向样品中加入20微升水,吹吸混匀后将样品置于恒温混匀仪上66℃,1400转/分钟,二十分钟,洗脱dna两次。进行pcr扩增。pcr结束后,向150微升的样品中加入72微升(1:0.48)ampurexp磁珠,用移液器吹吸混匀后,在旋转混匀仪上室温结合5分钟。将上清转移到新的低吸附ep管中,再加入78微升(1:1)ampurexp磁珠,混匀后在旋转混匀仪上室温结合5分钟,弃上清。用75%无水乙醇清洗磁珠两次,晾干后,向ep管中加入50微升水洗脱,从而得到大小在200碱基对到1000碱基对范围内的dna片段。经过上述步骤得到的文库(在本申请中,发明人将上述方法获得文库称为sishi-c文库)即可用于二代测序。
实施例2
为了对比本申请的sishi-c和前人的方法(raoetal.a3dmapofthehumangenomeatkilobaseresolutionrevealsprinciplesofchromatinlooping.cell.2014)在少量细胞研究中的效果,发明人利用口吸管精确地数出四组小鼠胚胎干细胞。每组细胞数目为500个。然后发明人分别利用sishi-c和前人的方法各对两组细胞(分别标记为重复试验1和重复试验2)进行建库。在pcr前测量dna的浓度,并在文库测序后分析有效的数据所占的比例。图8a结果显示,在pcr前,使用sishi-c方法的两组dna浓度均明显高于使用前人方法的两组;图8b结果显示,sishi-c显著减少了dna的损失,在测序数据中pcr产生的完全相同的序列所占的比例很少,有效的数据所占的比例明显高于使用前人的方法的两组。综上,和前人的方法相比,本申请的sishi-c显著地减少了dna在实验过程中的损失,非常适用于研究极少量细胞的基因组三维结构。
此外,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者更多个该特征。在本发明的描述中,“多个”的含义是两个或两个以上,除非另有明确具体的限定。
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
- 还没有人留言评论。精彩留言会获得点赞!