一种氨基聚乙二醇丙酸的制备方法与流程
本发明属于药物合成领域,具体涉及一种氨基聚乙二醇丙酸的制备方法。
背景技术:
聚乙二醇(polyethyleneglycol,peg)是由环氧乙烷与水或乙二醇逐步发生加成聚合而得到的一类分子量较低的水溶性聚醚。小分子量的低聚聚乙二醇为无色、无臭有吸湿性的粘稠液体,分子中既有醚链,又有羟基,故具有独特的溶解性能,能与水、醇混溶,微溶于醚。聚乙二醇(polyethyleneglycol,peg)近年来已广泛用于修饰生物大分子及小分子药物的结构。peg修饰生物大分子或小分子药物后,能显著改善其水溶性和稳定性,减少肾脏滤过效应,从而延长药物在生物体内的循环时间。
单分散长链聚乙二醇聚合度一定,分子量一定,结构一定,在医药、材料和工程等领域具有很重要的应用前景。例如,在药物制剂领域,聚乙二醇化的药物、多肽以及蛋白质等可以对药物的稳定性,免疫原性及药代动力学性质产生显著的影响。
聚乙二醇含两个活性端基,用于化学修饰时容易引起不期望的交联,因而如何高效合成单分散的小分子peg仍是当下一个难以攻克的难题。目前市面上的合成路线中普遍存在以下几个难点:
1)原料用量大,合成步骤多,产率低;
2)后处理困难,产品纯化难。
wu,xuan;zong,xi;ji,min等人公开了一种氨基-peg4-羧基的制备方法(journalofchemicalresearch,2016,vol.40,p.368-370),该方法以四甘醇为原料,经过加成,对甲基苯基磺酰化,叠氮化,还原等7步骤得到产品。该方法步骤多,产率较低,并且用到了危险的化学品叠氮化钠,raneyni等,不利于生产。反应路线如下:
技术实现要素:
本发明提供了一种氨基聚乙二醇羧基的制备方法,该制备方法优化了氨基聚乙二醇羧基的合成路线,该制备方法原料简单易得,步骤较短,纯化简单,无需柱层析分离纯化;不仅使得产品的提纯变得更为简单,还极大地提高了氨基聚乙二醇羧基的收率和纯度。
一种氨基聚乙二醇丙酸的制备方法,所述制备方法包括以下步骤:1)将双羟基聚乙二醇和金属碱溶于有机溶剂中,滴入丙烯酸叔丁酯,反应得到羟基聚乙二醇丙酸叔丁酯;2)将羟基聚乙二醇丙酸叔丁酯和碱溶于有机溶剂中,滴加对甲苯磺酰氯,反应得到对甲苯磺酸酯聚乙二醇丙酸叔丁酯;3)将对甲苯磺酸酯聚乙二醇丙酸叔丁酯溶于有机溶剂中,加入氢氧化铵与催化剂,反应得到氨基聚乙二醇丙酸叔丁酯;4)将氨基聚乙二醇丙酸叔丁酯溶于有机溶剂中,加入酸反应得到氨基聚乙二醇丙酸,所述双羟基聚乙二醇的结构式为:
进一步地,所述步骤1)中的有机溶剂为四氢呋喃、二氯甲烷、二氧六环中的一种。
进一步地,所述步骤1)中的金属碱为钠、钾、锂中的一种。
进一步地,所述步骤1)中的双羟基聚乙二醇、丙烯酸叔丁酯和金属碱的摩尔比为1:0.72~0.8:0.02~0.03。
进一步地,所述步骤2)中的碱为三乙胺、氢氧化钠、吡啶中的一种。
进一步地,所述步骤2)中的羟基聚乙二醇丙酸叔丁酯、对甲苯磺酰氯和碱的摩尔比为1:0.72~1.11:0.86~1.3。
进一步地,所述步骤2)中由羟基聚乙二醇丙酸制得对甲苯磺酸酯聚乙二醇丙酸叔丁酯的过程是于反应温度为20℃~40℃,反应时间为10~15h的条件下进行的。
进一步地,所述步骤3)中的催化剂为苄基三乙基氯化铵、四丁基溴化铵、四丁基氯化铵、四丁基硫酸氢铵中的一种。
进一步地,所述步骤3)中由对甲苯磺酸酯聚乙二醇丙酸叔丁酯制得氨基聚乙二醇丙酸叔丁酯的过程是于反应是于温度为20~50℃,反应时间为24~48h的条件下进行的。
进一步地,所述步骤4)中的酸为盐酸或三氟乙酸中的一种。
进一步地,所述步骤4)中由氨基聚乙二醇丙酸叔丁酯制得氨基聚乙二醇丙酸的过程是于反应的温度为20~80℃,反应时间为8~16h的条件下进行的。
该制备方法的工艺路线如下:
同现有技术相比,本发明的有益效果体现为:
1.原料简单易得,步骤较短,纯化简单,无需柱层析分离纯化,大大降低了纯化难度;
2.获得均一稳定的产物;
3.制备过程中,不涉及剧毒试剂,对环境污染低,绿色环保;
4.设备要求低,操作简单,步骤少,危险系数低,适合工业化大规模生产。
附图说明
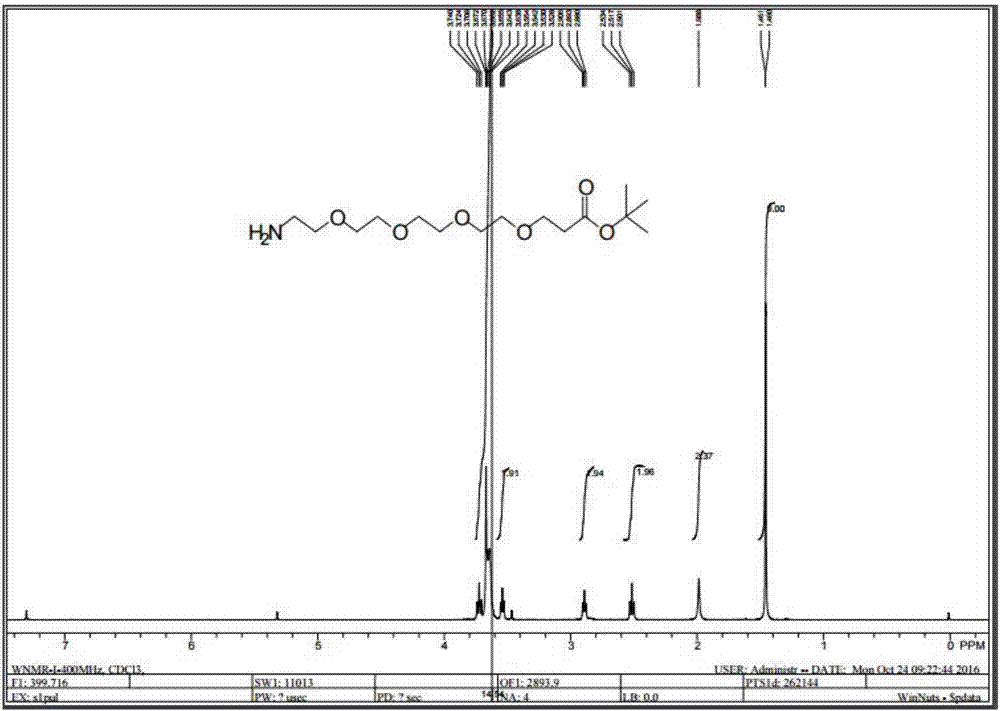
图1为实施例1的步骤(3)得到的中间产物的核磁氢谱图;
图2为实施例1的步骤(4)得到的纯产品的核磁氢谱图。
具体实施步骤
实施例1
(1)将100g四甘醇加入至500ml四氢呋喃中,称取0.36g碎的金属钠加入上述溶液中,并室温搅拌1h。称取53g丙烯酸叔丁酯滴加到上述反应液中,室温搅拌12h。tlc显示反应结束,加20ml水淬灭反应,旋除溶剂四氢呋喃,再加入300ml水,然后用400ml二氯甲烷萃取3次。无水硫酸钠干燥二氯甲烷相,旋干得到约100g粗产物;
(2)将步骤(1)得到的100g粗产物,40g三乙胺加入到400ml二氯甲烷中,搅拌,称取65g对甲苯磺酰氯溶于150ml二氯甲烷,在冰水浴条件下滴加至反应体系中,滴毕,20℃反应15h。tlc显示反应结束,加300ml水洗,分液,无水硫酸钠干燥有机相,旋干,得到约150g磺酰化的粗产物;
(3)将步骤(2)得到的150g磺酰化的粗产物加入到200ml二氯甲烷中,再加入100g氢氧化铵,10g四丁基溴化铵,升温到40℃,搅拌24h。反应结束,旋干,加入3m盐酸,调ph=3,用二氯甲烷300ml,萃洗2次,然后用氢氧化钠溶液调ph=9,再用二氯甲烷400ml萃取3次,无水硫酸钠干燥,旋干,得到50g氨基端的产物。核磁数据如下:1hnmr(400mhz,cdcl3):δ:3.724(t,j=6.4hz,2h);3.672~3.638(m,14h);3.530(t,j=4.8hz,2h);2.893(t,j=6.8hz,2h);1.989(s,2h);1.460(s,9h);
(4)将步骤(3)得到的50g氨基端的产物加入到100ml3m盐酸中,升温到60℃,搅拌8h。反应结束,调ph中性,然后旋干,抽滤,无水硫酸钠干燥滤液,旋干,得到35g纯品产物。核磁数据如下:1hnmr(400mhz,cdcl3):δ:3.775(t,j=4.8hz,2h);3.708~3.608(m,14h);3.167(t,j=4.2hz,2h);2.435(t,j=5.6hz,2h)。
实施例2
(1)将50g八甘醇加入至200ml二氯甲烷中,称取0.09g碎的金属钠加入上述溶液中,并室温搅拌1h。称取13g丙烯酸叔丁酯滴加到上述反应液中,室温搅拌12h。tlc显示反应结束,加20ml水淬灭反应,旋除溶剂二氯甲烷,再加入300ml水,然后用400ml二氯甲烷萃取3次。无水硫酸钠干燥二氯甲烷相,旋干得到约50g粗产物;
(2)将步骤(1)得到的50g粗产物,10g三乙胺加入到150ml二氯甲烷中,搅拌,称取16g对甲苯磺酰氯溶于50ml二氯甲烷,在冰水浴条件下滴加至反应体系中,滴毕,30℃反应12h。tlc显示反应结束,加100ml水洗,分液,无水硫酸钠干燥有机相,旋干,得到约60g磺酰化的粗产物;
(3)将步骤(2)得到的60g磺酰化的粗产物加入到150ml二氯甲烷中,再加入50g氢氧化铵,5g苄基三乙基氯化铵,升温到40℃,搅拌24h。反应结束,旋干,加入3m盐酸,调ph=3,用二氯甲烷100ml,萃洗2次,然后用氢氧化钠溶液调ph=9,再用二氯甲烷150ml萃取3次,无水硫酸钠干燥,旋干,得到30g氨基端的产物。核磁数据如下:1hnmr(400mhz,cdcl3):δ:3.736(t,j=6.4hz,2h);3.662~3.630(m,30h);3.535(t,j=4.8hz,2h);2.891(t,j=6.8hz,2h);1.987(s,2h);1.450(s,9h);
(4)将步骤(3)得到的30g氨基端的产物加入到100ml2m盐酸中,升温到60℃,搅拌8h。反应结束,调ph中性,然后旋干,抽滤,无水硫酸钠干燥滤液,旋干,得到25g纯品产物。核磁数据如下:1hnmr(400mhz,cdcl3):δ:3.777(t,j=4.8hz,2h);3.718~3.614(m,30h);3.161(t,j=4.2hz,2h);2.430(t,j=5.6hz,2h)。
实施例3
(1)将50g十二甘醇加入至200ml二氧六环中,称取0.06g碎的金属钠加入上述溶液中,并室温搅拌1h。称取8.7g丙烯酸叔丁酯滴加到上述反应液中,室温搅拌12h。tlc显示反应结束,加20ml水淬灭反应,旋除溶剂二氧六环,再加入300ml水,然后用400ml二氯甲烷萃取3次。无水硫酸钠干燥二氯甲烷相,旋干得到约50g粗产物;
(2)将步骤(1)得到的50g粗产物,3g氢氧化钠加入到150ml二氯甲烷中,搅拌,称取10.7g对甲苯磺酰氯溶于50ml二氯甲烷,在冰水浴条件下滴加至反应体系中,滴毕,40℃反应10h。tlc显示反应结束,加100ml水洗,分液,无水硫酸钠干燥有机相,旋干,得到约60g磺酰化的粗产物;
(3)将步骤(2)得到的60g磺酰化的粗产物加入到150ml二氯甲烷中,再加入50g氢氧化铵,5g四丁基氯化铵,升温到40℃,搅拌24h。反应结束,旋干,加入3m盐酸,调ph=3,用二氯甲烷100ml,萃洗2次,然后用氢氧化钠溶液调ph=9,再用二氯甲烷150ml萃取3次,无水硫酸钠干燥,旋干,得到40g氨基端的产物。核磁数据如下:1hnmr(400mhz,cdcl3):δ:3.722(t,j=6.4hz,2h);3.671~3.635(m,46h);3.528(t,j=4.8hz,2h);2.871(t,j=6.8hz,2h);1.995(s,2h);1.444(s,9h);
(4)将步骤(3)得到的30g氨基端的产物加入到100ml2m盐酸中,升温到60℃,搅拌8h。反应结束,调ph中性,然后旋干,抽滤,无水硫酸钠干燥滤液,旋干,得到25g纯品产物。核磁数据如下:1hnmr(400mhz,cdcl3):δ:3.768(t,j=4.8hz,2h);3.708~3.621(m,46h);3.151(t,j=4.2hz,2h);2.411(t,j=5.6hz,2h)。
实施例4
(1)将50g十六甘醇加入至200ml四氢呋喃中,称取0.05g碎的金属钾加入上述溶液中,并室温搅拌1h。称取6.5g丙烯酸叔丁酯滴加到上述反应液中,室温搅拌12h。tlc显示反应结束,加20ml水淬灭反应,旋除溶剂四氢呋喃,再加入300ml水,然后用400ml二氯甲烷萃取3次。无水硫酸钠干燥二氯甲烷相,旋干得到50g粗产物;
(2)将步骤(1)得到的50g粗产物,5g吡啶加入到150ml二氯甲烷中,搅拌,称取8g对甲苯磺酰氯溶于50ml二氯甲烷,在冰水浴条件下滴加至反应体系中,滴毕,30℃反应12h。tlc显示反应结束,加100ml水洗,分液,无水硫酸钠干燥有机相,旋干,得到约55g磺酰化的粗产物;
(3)将步骤(2)得到的55g磺酰化的粗产物加入到150ml甲醇中,再加入50g氢氧化铵,5g四丁基硫酸氢铵,升温到40℃,搅拌24h。反应结束,旋干,加入3m盐酸,调ph=3,用二氯甲烷100ml,萃洗2次,然后用氢氧化钠溶液调ph=9,再用二氯甲烷150ml萃取3次,无水硫酸钠干燥,旋干,得到42g氨基端的产物。核磁数据如下:1hnmr(400mhz,cdcl3):δ:3.711(t,j=6.4hz,2h);3.681~3.629(m,62h);3.525(t,j=4.8hz,2h);2.868(t,j=6.8hz,2h);2.052(s,2h);1.415(s,9h);
(4)将步骤(3)得到的42g氨基端的产物加入到150ml2.5m盐酸中,升温到60℃,搅拌8h。反应结束,调ph中性,然后旋干,抽滤,无水硫酸钠干燥滤液,旋干,得到30g纯品产物。核磁数据如下:1hnmr(400mhz,cdcl3):δ:3.760(t,j=4.8hz,2h);3.728~3.614(m,62h);3.143(t,j=4.2hz,2h);2.402(t,j=5.6hz,2h)。
实施例5
(1)将50g二十四甘醇加入至200ml四氢呋喃中,称取0.01g碎的金属锂加入上述溶液中,并室温搅拌1h。称取4.3g丙烯酸叔丁酯滴加到上述反应液中,室温搅拌12h。tlc显示反应结束,加20ml水淬灭反应,旋除溶剂四氢呋喃,再加入300ml水,然后用400ml二氯甲烷萃取3次。无水硫酸钠干燥二氯甲烷相,旋干得到约46g粗产物;
(2)将步骤(1)得到的46g粗产物,3.3g三乙胺加入到150ml二氯甲烷中,搅拌,称取5.3g对甲苯磺酰氯溶于30ml二氯甲烷,在冰水浴条件下滴加至反应体系中,滴毕,30℃反应12h。tlc显示反应结束,加100ml水洗,分液,无水硫酸钠干燥有机相,旋干,得到约48g磺酰化的粗产物;
(3)将步骤(2)得到的48g磺酰化的粗产物加入到150ml二氯甲烷中,再加入50g氢氧化铵,5g四丁基溴化铵,升温到40℃,搅拌24h。反应结束,旋干,加入3m盐酸,调ph=3,用二氯甲烷100ml,萃洗2次,然后用氢氧化钠溶液调ph=9,再用二氯甲烷150ml萃取3次,无水硫酸钠干燥,旋干,得到40g氨基端的产物。核磁数据如下:1hnmr(400mhz,cdcl3):δ:3.725(t,j=6.4hz,2h);3.657~3.622(m,94h);3.527(t,j=4.8hz,2h);2.886(t,j=6.8hz,2h);1.998(s,2h);1.436(s,9h);
(4)将步骤(3)得到的40g氨基端的产物加入到100ml2m三氟乙酸中,升温到60℃,搅拌8h。反应结束,调ph中性,然后旋干,抽滤,无水硫酸钠干燥滤液,旋干,得到33g纯品产物。核磁数据如下:1hnmr(400mhz,cdcl3):δ:3.766(t,j=4.8hz,2h);3.710~3.609(m,94h);3.155(t,j=4.2hz,2h);2.456(t,j=5.6hz,2h)。
- 还没有人留言评论。精彩留言会获得点赞!