一种透明、高耐热环烯烃共聚物及其制备方法与流程
本发明高分子材料领域,尤其涉及一种透明、高耐热环烯烃共聚物及其制备方法。
背景技术:
环烯烃共聚物(coc)是一种环烯烃结构的非晶型透明共聚高分子,有着和pmma匹敌的光学性能,比pmma和pc尺寸更稳定,耐热性比pc还高。coc有着低介电常数(绝缘性),玻璃化温度可调整性,透光性大于91%,耐热性具有较佳的耐热温度和抗氧化特性,热分解温度高于400度,并有良好的生物相容性和高流动性。coc材料使用无毒性单体为原料,聚合物纯度极高,水透过性非常低,无细胞毒素,无诱导有机体突变,无刺激性,符合fda标准。因此,环烯烃共聚物广泛地应用于制造各种光学、信息、电器、医用材料等。
环烯烃共聚物的耐热性能和热稳定性是这种材料的重要性能。在某些较高温度的使用环境下,如果环烯烃共聚物的耐热性能较差,则环烯烃共聚物会发生扭曲与变形等尺寸上的变化,从而直接影响环烯烃共聚物的光学性能和力学性能。因此,提高环烯烃共聚物的耐热性能和热稳定性能可大大扩展环烯烃共聚物的使用范围,提供耐热性以及热稳定性好的环烯烃共聚物是目前研究的重要课题。
技术实现要素:
有鉴于此,本发明所要解决的技术问题在于提供一种透明、高耐热环烯烃共聚物及其制备方法,本发明提供的环烯烃共聚物不仅具有好的耐热性能,而且具有好的热稳定性。
本发明提供了一种环烯烃共聚物,具有式(i)所示结构,
其中,140≤m≤220,170≤n≤280,1≤x≤4。
优选的,所述m为170≤m≤190。
优选的,所述n为170≤n≤190。
优选的,所述x为2≤x≤3。
本发明还提供了一种环烯烃共聚物的制备方法,包括:
1)将具有式(ii)结构的化合物和式(iii)结构的化合物在式(iv)结构的催化剂作用下进行聚合反应,得到聚合物;
其中,x为1≤x≤4;
2)将得到的聚合物用非均相金属催化剂进行氢化还原反应,得到环烯烃共聚物。
优选的,所述式(ii)结构的化合物和所述式(iii)结构的化合物的摩尔比为(9~1)∶1。
优选的,所述聚合反应的溶剂为c1~c15的烷烃、c1~c15的卤代烃、c3~c15的环烷烃或c5~c20的芳烃。
优选的,所述聚合反应的温度为10~50℃,所述聚合反应的时间为4~12h。
优选的,所述式(ii)结构的化合物和式(iii)结构的化合物的总摩尔数与所述式(iv)结构的催化剂的摩尔比为(25~1000)∶1。
优选的,所述非均相金属催化剂为5~10%湿钯碳、10%pd/c、pd/c/baso4或pt/sio2。
与现有技术相比,本发明提供了一种透明、高耐热环烯烃共聚物及其制备方法,本发明提供的环烯烃共聚物通过选择特定的结构,使得本发明提供的环烯烃共聚物玻璃化转变温度精准可调,范围为105-169℃,溶液涂膜,材料透光率可达91%,拉伸测试表明该材料具有优良的机械性能,拉伸强度为25-50mpa之间,拉伸模量为1600-2800mpa,断裂伸长率在2.5-5.4%之间,且10%热分解温度均大于410℃,具有很好的热稳定性。
此外,本发明还提供了一种所述透明、高耐热环烯烃共聚物环烯烃共聚物的制备方法,本发明提供的制备方法通过采用具有式(iv)结构的苯亚甲基·二氯·双(三环己基膦)合钌配合物催化具有式(ii)结构的多元环状环烯单体(tcpd)和式(iii)结构的带有短链酯键的降冰片烯单体进行无规共聚合,不仅引发速率快,催化转化率达到100%,且不发生交联等副反应;相较于结构不明确的w系,mo系催化剂,本发明的聚合方法不需要助催化剂,聚合耐受性很好,分子量分布可调。对所得开环易位聚合物进行氢化还原,可得到主链完全饱和的聚合物。
附图说明
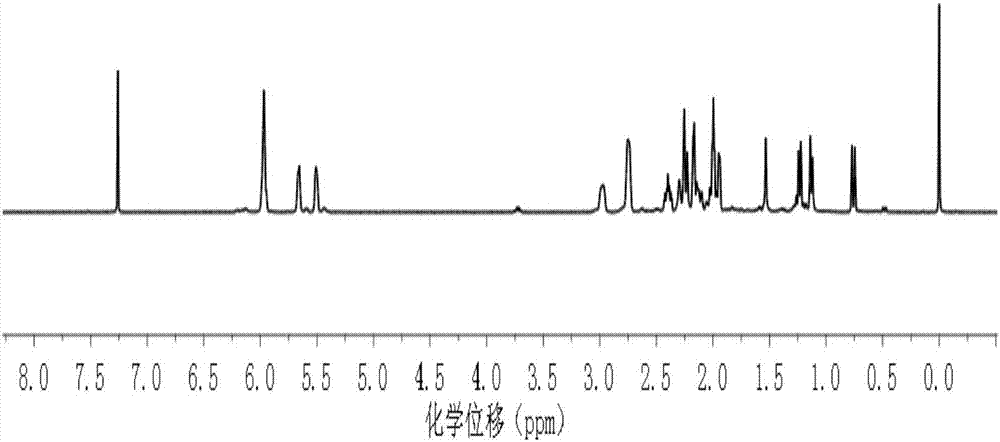
图1为本发明实施例1得到的产品的核磁共振氢谱图;
图2为本发明实施例2得到的产品的核磁共振氢谱图;
图3为本发明实施例3得到的聚合反应产物氢化前后的核磁共振氢谱图;
图4为本发明实施例3,实施例4,实施例5,实施例6和实施例7得到的环烯烃共聚物的示差扫描量热曲线;
图5为本发明实施例3,实施例4,实施例5,实施例6和实施例7得到环烯烃共聚物在氮气中的热失重曲线;
图6为本发明实施例3、实施例5和实施例7得到的环烯烃共聚物的透光率;
图7为本发明实施例4得到的聚合反应产物氢化前后的核磁共振氢谱图;
图8为本发明实施例5得到的聚合反应产物氢化前后的核磁共振氢谱图;
图9为本发明实施例6得到的聚合反应产物氢化前后的核磁共振氢谱图;
图10为本发明实施例7得到的聚合反应产物氢化前后的核磁共振氢谱图。
具体实施方式
本发明提供了一种环烯烃共聚物,具有式(i)所示结构,
其中,所述140≤m≤220,170≤n≤280,1≤x≤4。
按照本发明,所述m优选为170≤m≤190。所述n优选为170≤n≤190。所述x优选为1≤x≤2,更优选为1;所述环烯烃共聚物为无规共聚物;所述共聚物重均分子量为7.0~15.0×104g/mol,分子量分布指数为1.4~2.0,玻璃化转变温度为105~169℃。
本发明提供的环烯烃共聚物通过选择特定的结构,使得本发明提供的环烯烃共聚物玻璃化转变温度精准可调,范围在105~169℃之间,环烯烃聚合物薄膜厚度为50~120μm时透光率大于91%,机械性能优良,拉伸强度在25~50mpa之间,拉伸模量为1600~2800mpa,断裂伸长率在2.5~5.4%之间,且10%热分解温度均大于410℃,具有很好的热稳定性。
本发明还提供了一种环烯烃共聚物的制备方法,包括:
1)将具有式(ii)结构的化合物和式(iii)结构的化合物在式(iv)结构的催化剂作用下进行聚合反应,得到聚合物;
其中,x为1≤x≤4;
2)将得到的聚合物用非均相金属催化剂进行氢化还原,得到环烯烃共聚物。
按照本发明,本发明将具有式(ii)结构的化合物和式(iii)结构的化合物在式(iv)结构的催化剂作用下进行聚合反应,得到聚合物;所述式(ii)结构的化合物和所述式(iii)结构的化合物的摩尔比优选为(9~1)∶1,更优选为更优选为(7~2)∶1,最优选为(6~3)∶1;所述式(ii)结构的化合物和式(iii)结构的化合物的总摩尔数与所述式(iv)结构的催化剂的摩尔比优选为(25~1000)∶1,更优选为(50~100)∶1;所述聚合反应的温度优选为10~50℃,更优选为25~30℃;所述聚合反应的时间优选为4~12h,更优选为7~9小时;所述聚合反应还加入链终止剂终止反应,所述终止剂优选为乙烯基乙醚,所述终止剂的摩尔数优选为式(iv)结构的催化剂摩尔数的100~500倍,更优选为300~400倍。
具体的,本发明优选首先具有式(ii)结构的化合物和式(iii)结构的化合物和溶剂混合;其中,所述的聚合物浓溶液浓度优选为5~15wt%,更优选为8~10wt%。本发明对混合的方式没有特要求,本领域公知的混合方式均可,其中,所述混合时搅拌的时间优选为5分钟~15分钟,更优选为8分钟~12分钟,最优选为10分钟。所述溶剂优选为所述聚合反应的溶剂优选为c1~c15的烷烃、c1~c15的卤代烃、c3~c15的环烷烃或c5~c20的芳烃,更优选为c5~c8的烷烃、c1~c5的卤代烃、c5~c8的环烷烃或c6~c15的芳烃,最优选为二氯甲烷;本发明对溶剂的来源没有特殊要求,仅需无水无空气的溶剂即可,所述无水无空气溶剂的处理方式优选采用液氮冷冻~融化处理方式进行处理。
本发明所述聚合反应优选在干燥、无氧条件下进行,本发明对聚合反应的装置没有特殊要求,可以在布劳恩(mbraun)手套箱中进行,也可以采用标准的希莱克(schlenk)技术在氮气的保护下进行。
按照本发明,所述聚合反应完成后,本发明优选采用停止搅拌终止所述聚合反应,得到聚合反应溶液;将所述聚合反应溶液和沉淀剂混合,得到沉淀产物;将所述沉淀产物过滤、洗涤、干燥,得到聚合反应产物。其中,所述沉淀剂优选为无水甲醇;本发明对所述沉淀产物过滤、洗涤和干燥的方法没有特殊的限制,采用本领域技术人员熟知的过滤、洗涤和干燥的技术方案即可。在本发明中,所述沉淀产物洗涤的试剂优选为乙醇。在本发明中,所述沉淀产物洗涤的次数优选为2次~4次,更优选为3次。在本发明中,所述沉淀产物干燥的方法优选为真空干燥。在本发明中,所述沉淀产物干燥的温度优选为20℃~40℃,更优选为25℃~35℃,最优选为30℃。在本发明中,所述沉淀产物干燥的时间优选为12小时~24小时,更优选为16小时~20小时,最优选为18小时。
本发明对所述具有式ii和iii结构的化合物的来源没有特殊的限制,采用本领域技术人员熟知的制备具有式ii和iii结构化合物的方法制备得到即可。在本发明中,所述具有式ii结构的化合物的制备方法优选为:
将双环戊二烯和2,6-二叔丁基对甲基苯酚进行反应,得到具有式ii结构的化合物。其中,所述双环戊二烯和2,6-二叔丁基对甲基苯酚的摩尔比优选为(800~1200)∶1,更优选为(900~1100)∶1,最优选为(1000~1100)∶1。所述反应的氛围优选为真空或氮气氛围;所述反应的温度优选为180℃~220℃,更优选为190℃~210℃,最优选为200℃。所述反应的时间优选为14小时~18小时,更优选为15小时~17小时。
所述双环戊二烯和2,6-二叔丁基对甲基苯酚反应完成后,本发明优选将得到的双环戊二烯和2,6-二叔丁基对甲基苯酚反应物冷却、减压蒸馏、向残渣加入醇、过滤除不溶物、低温重结晶,析出固体后再次用醇低温重结晶,得到具有式ii结构的化合物。本发明对所述冷却、减压蒸馏、过滤和低温重结晶的方法没有特殊的限制,采用本领域技术人员熟知的冷却、减压蒸馏、过滤和低温重结晶技术方案即可。在本发明中,所述冷却的温度优选为20℃~30℃,更优选为24℃~28℃。在本发明中,所述减压蒸馏的温度优选为40℃~80℃,更优选为50℃~60℃。在本发明中,所述低温重结晶的温度优选为~20℃~0℃,更优选为~10℃~~5℃。
本发明对所述具有式iii结构化合物的来源没有特殊的限制,采用本领域技术人员熟知的制备具有式iii结构化合物的方法制备得到即可。在本发明中,所述具有式iii结构的化合物的制备方法优选为:
将双环戊二烯、丙烯酸甲酯和2,6-二叔丁基对甲基苯酚进行反应,得到具有式iii结构的化合物;所述反应优选在真空或氮气保护氛围下进行;所述反应的温度优选为180℃~220℃,更优选为190℃~210℃,最优选为200℃。在本发明中,所述反应的时间优选为4小时~8小时,更优选为5小时~7小时,最优选为6小时。
所述双环戊二烯、丙烯酸甲酯和2,6-二叔丁基对甲基苯酚反应完成后,本发明优选将得到的双环戊二烯、丙烯酸甲酯和2,6-二叔丁基对甲基苯酚反应产物冷却、静置、减压蒸馏,得到具有式iii结构的化合物。本发明对所述冷却、静置和减压蒸馏的方法没有特殊的限制,采用本领域技术人员熟知的冷却、静置和减压蒸馏的技术方案即可。在本发明中,所述冷却的温度优选为20℃~30℃,更优选为25℃~28℃。在本发明中,所述静置的时间优选为10小时~16小时,更优选为12小时~14小时。在本发明中,所述减压蒸馏的温度优选为110℃~160℃,更优选为120℃~150℃。本发明优选收集所述减压蒸馏时130℃~140℃的馏分,所述馏分即为具有式iii结构的化合物。
按照本发明,本发明还将得到的聚合物用非均相金属催化剂进行氢化还原,得到环烯烃共聚物。其中,所述非均相金属催化剂优选为5%~10%湿钯碳;本所所述的非均相金属催化剂与所述化聚合物质量比优选为(3~6)∶1,更优选为(5~6)∶1;所述反应的溶剂优选为环己烷;所述反应优选还加入少量助催化剂bht;所述反应中氢气的压力优选为5.5~6.0mpa,更优选为5.8~5.9mpa;所述反应的温度优选为120~130℃;所述反应的时间优选为22~24小时。反应完毕后,本发明优选将反应液缓慢倒入工业乙醇中,析出大量白色固体,过滤,重复洗涤三次后放入真空烘箱中60℃下干燥12~24小时即得环烯烃共聚物。
更具体的,本发明所述环烯烃共聚物(coc)的制备可按照下述反应式进行:
本发明提供的所述环烯烃共聚物的制备方法,通过采用具有式(iv)结构的苯亚甲基·二氯·双(三环己基膦)合钌配合物催化具有式(ii)结构的多元环状环烯单体(tcpd)和式(iii)结构的带有短链酯键的降冰片烯单体(nbma)进行无规共聚合,不仅引发速率快,催化转化率达到100%,且不发生交联等副反应;相较于结构不明确的w系,mo系催化剂,本发明的聚合方法不需要助催化剂,聚合耐受性很好,分子量分布可调。对所得开环易位聚合物进行氢化还原,可得到主链完全饱和的聚合物。且本发明可通过控制所述聚合反应原料的用量控制制备得到的环烯烃共聚物的玻璃化转变温度。本发明制备得到的环烯烃共聚物中具有式ii结构的化合物越多,环烯烃共聚物的玻璃化转变温度越高。
下面将结合本发明实施例的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
以下实施例中二氯甲烷、甲苯等无水无氧溶剂由mbraunsps溶剂纯化处理系统制得;所采用的grubbs第一代催化剂参照文献(jacs,1996,118,100~110)描述的方法制备,多元状烯单体(tcpd)的制备按照文献(2010wileyperiodicals,inc.jpolymsciparta:polymchem49:938-944,2011)描述的方法制备。其他原料均为市售产品。
本申请在对制备的高性能环烯烃共聚物进行检测的过程中,采用核磁共振波谱测定共聚物的分子结构;采用差热分析法测定聚合物的熔融温度;采用凝胶渗透色谱测定聚合物的分子量与分子量分布指数。其中,核磁共振波谱是指聚合物的1h和13cnmr谱由varianunity~400型核磁共振波谱仪在25℃下测定,tms为内标,氘代氯仿为溶剂。示差热分析(dsc)是指聚合物的玻璃化转变温度由perkin~elmerpyrisldsc示差扫描量热仪测定,升降温速率为20℃/min,二次扫描。热失重(tga)由perkin-elmerpyris1型仪器测定。透光率利用shimadzuuv~3600紫外~可见光~近红外分光光度计测定,测定波长为400~800nm。拉伸实验:聚合物的力学性能表征是在instron1121,canton,ma仪器上进行的,以gb/t1040~1992为标准,样条夹距为20.0mm,测试速率为5mm/min,每个样品至少测试8遍以保证数据的可靠性。凝胶渗透色谱(gpc)是指聚合物的分子量和分子量分布指数由waters1525型凝胶渗透色谱仪测定;采用ri~laser检测仪,,溶剂为四氢呋喃(thf),测试温度为35℃,流速为1.0ml/min,采用pleasicalps~1为标样。
实施例1
向2升的单口瓶中依次加入1.5升双环戊二烯,1克2,6-二叔丁基对甲基苯酚,对所述单口瓶重复进行3次抽真空后充氮气的操作;将所述单口瓶瓶加热至200℃,使所述单口瓶中的物质在搅拌的条件下进行16小时的反应。
所述反应结束后,将得到的反应产物冷却至25℃,减压蒸馏得到低沸点的未反应物1100克。向残渣内加入1升乙醇,过滤除不溶物后滤液冷冻3小时。重结晶后过滤得固体389克,再次用800毫升乙醇重结晶得固体315克。本发明实施例1提供的方法制备得到产品的产率为78%。
将上述得到的产品进行核磁共振氢谱检测,检测结果如图1,图1为本发明实施例1得到的产品的核磁共振氢谱图,由图1可知,本发明实施例1得到的产品为具有式ii结构的化合物。
实施例2
向1升的不锈钢高压釜中依次加入115克双环戊二烯,310克丙烯酸甲酯,1克的2,6-二叔丁基对甲基苯酚,对所述高压釜重复进行3次抽真空后充氮气的操作;将所述高压釜加热至200℃,使所述高压釜中的物质在搅拌的条件下进行16小时的反应。
反应结束后,将得到的反应产物冷却至25℃,静置12小时后在65℃下进行减压蒸馏,收集未反应的双环戊二烯和环戊二烯;将减压蒸馏后得到的产物在100℃下再次进行减压蒸馏,收集减压蒸馏时的馏分,得114克产品。本发明实施例2提供的方法制备得到产品的产率为70%。
将上述得到的产品进行核磁共振氢谱检测,检测结果如图2,图2为本发明实施例2得到的产品的核磁共振氢谱图,由图2可知,本发明实施例2得到的产品为具有式iii结构的化合物。
实施例3
室温下,在干燥的聚合反应瓶中,加入0.12gtcpd单体和0.01gnbma单体,加入50ml二氯甲烷,充分搅拌10min,称2.5mg(g1于小安瓶中,加入5ml二氯甲烷,超声3min,使充分溶解,导入到聚合瓶中,聚合开始,五分钟内,溶液逐渐由紫色变为橙黄色,继续搅拌7小时,加入乙烯基乙醚eve(500当量)终止聚合,搅拌半个小时。将反应液倒入无水甲醇中,析出大量白色聚合物。过滤得到的产品以丙酮洗涤3次后放入真空烘箱中40℃下干燥12小时。称重得0.13g聚合物,收率为100%。
在干燥的高压釜中,依次加入上述聚合物0.13g,5%湿钯碳0.52g(4eqv),少许bht,40ml环己烷溶剂,升压5.8mp,升温至130℃,搅拌24小时,过滤后将反应液缓慢滴加到100ml工业乙醇中,析出大量白色固体,过滤抽干,将所得白色固体重新用40ml甲苯加热溶解半个小时,后慢慢滴加到300ml工业乙醇中,过滤抽干,该过程重复2次后放入真空烘箱中,60℃下干燥12小时,得0.12g产品,产率92.3%,纯度大于99.9%。
对本发明实施例3得到的环烯烃共聚物进行核磁共振氢谱检测,检测结果如图3,图3为本发明实施例3得到的聚合反应产物和环烯烃共聚物的核磁共振氢谱图,图3中曲线1为本发明实施例3氢化前得到的聚合反应产物的核磁共振氢谱,曲线2为本发明实施例3氢化后得到的环烯烃共聚物的核磁共振氢谱,由图3可以看出,本发明实施例3得到的聚合反应产物经过氢化反应后双键峰完全消失,氢化效果较好。
对本发明实施例3得到的环烯烃共聚物进行凝胶渗透色谱测试,测试结果为本发明实施例3得到的环烯烃共聚物的分子量分布为1.4,重均分子量为7.0×104g/mol。
对本发明实施例3得到的环烯烃共聚物进行差热分析法测试,测试结果如图4,图4为本发明实施例3,实例4,实例5,实例6,实例7得到的环烯烃共聚物的示差扫描量热曲线,由图4中曲线1可知,本发明实施例3得到的环烯烃共聚物没有熔融温度,为非晶态,本发明实施例3得到的环烯烃共聚物的玻璃化转变温度为169℃。对本发明实施例3得到的环烯烃共聚物进行热失重法测试,测试结果如图5,图5为本发明实施例3,实例4,实例5,实例6,实例7得到的环烯烃共聚物在氮气中的热重曲线,图5中曲线1为本发明实施例3得到的环烯烃共聚物在氮气中的热重曲线;由图5可知,本发明实施例3得到的环烯烃共聚物在410℃时的分解率为10%,具有较高的热稳定性。
对本发明实施例3得到的环烯烃共聚物的力学性能进行测试,测试结果为本发明实施例3得到的环烯烃共聚物的断裂伸长率为2.5%,拉伸强度为25mpa,拉伸模量为1600mpa。
对本发明实施例3得到的环烯烃共聚物的透明性进行测试,测试结果如图6,图6为本发明实施例3、实施例5和实施例7得到的环烯烃共聚物的透光率,图6中曲线1为本发明实施例3得到的环烯烃共聚物的透光率,由图6可知,本发明实施例3得到的环烯烃共聚物的透光率>91%。
实施例4
室温下,在干燥的聚合反应瓶中,加入0.21gtcpd单体和0.04gnbma单体,加入50ml二氯甲烷,充分搅拌10min,称10mg(g1于小安瓶中,加入5ml二氯甲烷,超声3min,使充分溶解,导入到聚合瓶中,聚合开始,五分钟内,溶液逐渐由紫色变为橙黄色,继续搅拌7小时,加入乙烯基乙醚eve(500当量)终止聚合,搅拌半个小时。将反应液倒入无水甲醇中,析出大量白色聚合物。过滤得到的产品以丙酮洗涤3次后放入真空烘箱中40℃下干燥12小时。称重得0.25g聚合物,收率为100%。
在干燥的高压釜中,依次加入上述聚合物0.25g,5%湿钯碳1g(4eqv),少许bht,40ml环己烷溶剂,升压5.8mp,升温至130℃,搅拌24小时,过滤后将反应液缓慢滴加到100ml工业乙醇中,析出大量白色固体,过滤抽干,将所得白色固体重新用40ml甲苯加热溶解半个小时,后慢慢滴加到300ml工业乙醇中,过滤抽干,该过程重复2次后放入真空烘箱中,60℃下干燥12小时,得0.24g产品,产率96%,纯度大于99.9%。
对本发明实施例4得到的环烯烃共聚物进行核磁共振氢谱检测,检测结果如图7,图7为本发明实施例4得到的聚合反应产物和环烯烃共聚物的核磁共振氢谱图,图4中曲线1为本发明实施例4氢化前得到的聚合反应产物的核磁共振氢谱,曲线2为本发明实施例4氢化后得到的环烯烃共聚物的核磁共振氢谱,由图7可以看出,本发明实施例4得到的聚合反应产物经过氢化反应后双键峰完全消失,氢化效果较好。
对本发明实施例4得到的环烯烃共聚物进行凝胶渗透色谱测试,测试结果为本发明实施例4得到的环烯烃共聚物的分子量分布为1.4,重均分子量为9.4×104g/mol。
对本发明实施例4得到的环烯烃共聚物进行差热分析法测试,测试结果如图4,图4为本发明实施例3,实例4,实例5,实例6,实例7得到的环烯烃共聚物的示差扫描量热曲线,由图4中曲线2可知,本发明实施例4得到的环烯烃共聚物没有熔融温度,为非晶态,本发明实施例4得到的环烯烃共聚物的玻璃化转变温度为154℃。对本发明实施例4得到的环烯烃共聚物进行热失重法测试,测试结果如图5,图5为本发明实施例3,实例4,实例5,实例6,实例7得到的环烯烃共聚物在氮气中的热重曲线,图5中曲线2为本发明实施例4得到的环烯烃共聚物在氮气中的热重曲线;由图5可知,本发明实施例4得到的环烯烃共聚物在413℃时的分解率为10%,具有较高的热稳定性。
对本发明实施例4得到的环烯烃共聚物的力学性能进行测试,测试结果为本发明实施例4得到的环烯烃共聚物的断裂伸长率为2.8%,拉伸强度为32mpa,拉伸模量为2200mpa。
实施例5
室温下,在干燥的聚合反应瓶中,加入0.76gtcpd单体和0.25gnbma单体,加入50ml二氯甲烷,充分搅拌10min,称20mg(g1于小安瓶中,加入5ml二氯甲烷,超声3min,使充分溶解,导入到聚合瓶中,聚合开始,五分钟内,溶液逐渐由紫色变为橙黄色,继续搅拌7小时,加入乙烯基乙醚eve(500当量)终止聚合,搅拌半个小时。将反应液倒入无水甲醇中,析出大量白色聚合物。过滤得到的产品以丙酮洗涤3次后放入真空烘箱中40℃下干燥12小时。称重得1.01g聚合物,收率为100%。
在干燥的高压釜中,依次加入上述聚合物1.01g,5%湿钯碳4g(4eqv),少许bht,40ml环己烷溶剂,升压5.8mp,升温至130℃,搅拌24小时,过滤后将反应液缓慢滴加到100ml工业乙醇中,析出大量白色固体,过滤抽干,将所得白色固体重新用40ml甲苯加热溶解半个小时,后慢慢滴加到300ml工业乙醇中,过滤抽干,该过程重复2次后放入真空烘箱中,60℃下干燥12小时,得9.9g产品,产率98.1%,纯度大于99.9%。
对本发明实施例5得到的环烯烃共聚物进行核磁共振氢谱检测,检测结果如图8,图8为本发明实施例5得到的聚合反应产物和环烯烃共聚物的核磁共振氢谱图,图8中曲线1为本发明实施例5氢化前得到的聚合反应产物的核磁共振氢谱,曲线2为本发明实施例5氢化后得到的环烯烃共聚物的核磁共振氢谱,由图8可以看出,本发明实施例5得到的聚合反应产物经过氢化反应后双键峰完全消失,氢化效果较好。
对本发明实施例5得到的环烯烃共聚物进行凝胶渗透色谱测试,测试结果为本发明实施例5得到的环烯烃共聚物的分子量分布为1.4,重均分子量为11.8×104g/mol。
对本发明实施例5得到的环烯烃共聚物进行差热分析法测试,测试结果如图4,图4为本发明实施例3,实例4,实例5,实例6,实例7得到的环烯烃共聚物的示差扫描量热曲线,由图4中曲线1可知,本发明实施例3得到的环烯烃共聚物没有熔融温度,为非晶态,本发明实施例5得到的环烯烃共聚物的玻璃化转变温度为147℃。对本发明实施例5得到的环烯烃共聚物进行热失重法测试,测试结果如图5,图5为本发明实施例3,实例4,实例5,实例6,实例7得到的环烯烃共聚物在氮气中的热重曲线,图5中曲线3为本发明实施例5得到的环烯烃共聚物在氮气中的热重曲线;由图5可知,本发明实施例5得到的环烯烃共聚物在415℃时的分解率为10%,具有较高的热稳定性。
对本发明实施例5得到的环烯烃共聚物的力学性能进行测试,测试结果为本发明实施例5得到的环烯烃共聚物的断裂伸长率为3.1%,拉伸强度为36mpa,拉伸模量为2400mpa。
对本发明实施例5得到的环烯烃共聚物的透明性进行测试,测试结果如图6,图6为本发明实施例3、实施例5和实施例7得到的环烯烃共聚物的透光率,图6中曲线2为本发明实施例5得到的环烯烃共聚物的透光率,由图6可知,本发明实施例5得到的环烯烃共聚物的透光率>91%。
实施例6
室温下,在干燥的聚合反应瓶中,加入0.7gtcpd单体和0.3gnbma单体,加入50ml二氯甲烷,充分搅拌10min,称20mg(g1于小安瓶中,加入5ml二氯甲烷,超声3min,使充分溶解,导入到聚合瓶中,聚合开始,五分钟内,溶液逐渐由紫色变为橙黄色,继续搅拌7小时,加入乙烯基乙醚eve(500当量)终止聚合,搅拌半个小时。将反应液倒入无水甲醇中,析出大量白色聚合物。过滤得到的产品以丙酮洗涤3次后放入真空烘箱中40℃下干燥12小时。称重得1g聚合物,收率为100%。
在干燥的高压釜中,依次加入上述聚合物1g,5%湿钯碳4g(4eqv),少许bht,40ml环己烷溶剂,升压5.8mp,升温至130℃,搅拌24小时,过滤后将反应液缓慢滴加到100ml工业乙醇中,析出大量白色固体,过滤抽干,将所得白色固体重新用40ml甲苯加热溶解半个小时,后慢慢滴加到300ml工业乙醇中,过滤抽干,该过程重复2次后放入真空烘箱中,60℃下干燥12小时,得0.96g产品,产率96%,纯度大于99.9%。
对本发明实施例6得到的环烯烃共聚物进行核磁共振氢谱检测,检测结果如图9,图9为本发明实施例6得到的聚合反应产物和环烯烃共聚物的核磁共振氢谱图,图9中曲线1为本发明实施例6氢化前得到的聚合反应产物的核磁共振氢谱,曲线2为本发明实施例6氢化后得到的环烯烃共聚物的核磁共振氢谱,由图9可以看出,本发明实施例6得到的聚合反应产物经过氢化反应后双键峰完全消失,氢化效果较好。
对本发明实施例6得到的环烯烃共聚物进行凝胶渗透色谱测试,测试结果为本发明实施例6得到的环烯烃共聚物的分子量分布为1.4,重均分子量为13.9×104g/mol。
对本发明实施例6得到的环烯烃共聚物进行差热分析法测试,测试结果如图4,图4为本发明实施例3,实例4,实例5,实例6,实例7得到的环烯烃共聚物的示差扫描量热曲线,由图4中曲线4可知,本发明实施例6得到的环烯烃共聚物没有熔融温度,为非晶态,本发明实施例6得到的环烯烃共聚物的玻璃化转变温度为131℃。对本发明实施例6得到的环烯烃共聚物进行热失重法测试,测试结果如图5,图5为本发明实施例3,实例4,实例5,实例6,实例7得到的环烯烃共聚物在氮气中的热重曲线,图5中曲线4为本发明实施例6得到的环烯烃共聚物在氮气中的热重曲线;由图5可知,本发明实施例6得到的环烯烃共聚物在415℃时的分解率为10%,具有较高的热稳定性。
对本发明实施例6得到的环烯烃共聚物的力学性能进行测试,测试结果为本发明实施例6得到的环烯烃共聚物的断裂伸长率为3.7%,拉伸强度为402mpa,拉伸模量为2400mpa。
实施例7
室温下,在干燥的聚合反应瓶中,加入0.6gtcpd单体和0.44gnbma单体,加入50ml二氯甲烷,充分搅拌10min,称20mg(g1于小安瓶中,加入5ml二氯甲烷,超声3min,使充分溶解,导入到聚合瓶中,聚合开始,五分钟内,溶液逐渐由紫色变为橙黄色,继续搅拌7小时,加入乙烯基乙醚eve(500当量)终止聚合,搅拌半个小时。将反应液倒入无水甲醇中,析出大量白色聚合物。过滤得到的产品以丙酮洗涤3次后放入真空烘箱中40℃下干燥12小时。称重得1.04g聚合物,收率为100%。
在干燥的高压釜中,依次加入上述聚合物1.04g,5%湿钯碳4g(4eqv),少许bht,40ml环己烷溶剂,升压5.8mp,升温至130℃,搅拌24小时,过滤后将反应液缓慢滴加到100ml工业乙醇中,析出大量白色固体,过滤抽干,将所得白色固体重新用40ml甲苯加热溶解半个小时,后慢慢滴加到300ml工业乙醇中,过滤抽干,该过程重复2次后放入真空烘箱中,60℃下干燥12小时,得1g产品,产率96.1%,纯度大于99.9%。
对本发明实施例7得到的环烯烃共聚物进行核磁共振氢谱检测,检测结果如图10,图10为本发明实施例7得到的聚合反应产物和环烯烃共聚物的核磁共振氢谱图,图10中曲线1为本发明实施例7氢化前得到的聚合反应产物的核磁共振氢谱,曲线2为本发明实施例7氢化后得到的环烯烃共聚物的核磁共振氢谱,由图10可以看出,本发明实施例7得到的聚合反应产物经过氢化反应后双键峰完全消失,氢化效果较好。
对本发明实施例7得到的环烯烃共聚物进行凝胶渗透色谱测试,测试结果为本发明实施例7得到的环烯烃共聚物的分子量分布为2.0,重均分子量为15.5×104g/mol。
对本发明实施例7得到的环烯烃共聚物进行差热分析法测试,测试结果如图4,图4为本发明实施例3,实例4,实例5,实例6,实例7得到的环烯烃共聚物的示差扫描量热曲线,由图4中曲线5可知,本发明实施例7得到的环烯烃共聚物没有熔融温度,为非晶态,本发明实施例7得到的环烯烃共聚物的玻璃化转变温度为105℃。对本发明实施例5得到的环烯烃共聚物进行热失重法测试,测试结果如图5,图5为本发明实施例3,实例4,实例5,实例6,实例7得到的环烯烃共聚物在氮气中的热重曲线,图5中曲线5为本发明实施例7得到的环烯烃共聚物在氮气中的热重曲线;由图5可知,本发明实施例7得到的环烯烃共聚物在418℃时的分解率为10%,具有较高的热稳定性。
对本发明实施例7得到的环烯烃共聚物的力学性能进行测试,测试结果为本发明实施例7得到的环烯烃共聚物的断裂伸长率为5.4%,拉伸强度为50mpa,拉伸模量为2800mpa。
对本发明实施例7得到的环烯烃共聚物的透明性进行测试,测试结果如图6,图6为本发明实施例3、实施例5和实施例7得到的环烯烃共聚物的透光率,图6中曲线3为本发明实施例7得到的环烯烃共聚物的透光率,由图6可知,本发明实施例7得到的环烯烃共聚物的透光率>91%。
具体的,本发明实施例3~实施例7中投料反应条件见表1;
表1
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!