一种螺旋纳米碳纤维表面修饰纳米SiO2粒子的方法与流程
本发明属于无机材料制备领域,具体涉及一种螺旋纳米碳纤维表面修饰纳米SiO2粒子的方法。
技术背景
随着现代社会的发展,人们对环保的要求越来越高,在减少石化燃料使用的同时,又能带来更好的产品成为了一个亟待解决的问题。美国权威机构的测试表明,轮胎滚动阻力每下降3%~5%,汽车就可节约燃油1%,但同时实现耐磨性、抗湿滑性能和低滚动阻力三种性能的提高,是传统轮胎面临的技术瓶颈。根据现有文献报道表明,将螺旋纳米碳纤维作为新型填料添加到炭黑补强橡胶的体系中,可利用特殊的纳米螺旋结构提高橡胶复合材料的力学性能。在螺旋纳米碳纤维表面原位生长纳米SiO2粒子,不仅可以凭借其凸出于螺旋表面的纳米微粒结构缠绕更多橡胶分子链,而且还可发挥白炭黑在补强橡胶的抗湿滑路面牵引性优势,弥补碳填料补强橡胶的劣势。
目前,在纳米碳管或纳米碳纤维表面接枝生长SiO2粒子,通常需要对纳米碳管或纳米碳纤维进行表面基团接枝改性,工艺复杂,SiO2粒子的形貌及分布较难控制。例如,中国发明专利(201611063545.2)公开了一种SiO2/螺旋纳米碳纤维双相填料的制备方法,该方法主要包含四个步骤:(1)对螺旋纳米碳纤维进行酸化,得到羟基化螺旋纳米碳纤维;(2)将所述羟基化螺旋纳米碳纤维与聚丙烯酸、二环己基碳二亚胺、二甲氨基吡啶和有机溶剂混合进行接枝反应,得到聚丙烯酸接枝螺旋纳米碳纤维;(3)将所述聚丙烯酸接枝螺旋纳米碳纤维与二环己基碳二亚胺、二甲氨基吡啶和硅烷偶联剂混合进行硅烷化反应,得到硅氧烷接枝螺旋纳米碳纤维;(4)将所述硅氧烷接枝螺旋纳米碳纤维与正硅酸已酯、氨水和有机溶剂混合进行水解接枝反应,得到SiO2/螺旋纳米碳纤维双相填料。该专利技术存在以下不足:(a)螺旋纳米碳纤维的前处理过程(即官能团接枝改性,包括一个酸化改性和两个预接枝反应过程)工艺步骤繁琐,出现了三个中间产物,工艺可控性与实验结果可重复性较差;(b)使用了大量有机试剂,比如聚丙烯酸、二环己基碳二亚胺、二甲氨基吡啶、硅烷偶联剂、四氢呋喃、丙酮等,大大增加了制备成本;(c)螺旋纳米碳纤维的前处理工艺排放了较多废液,不够环保。
技术实现要素:
针对现有技术存在的上述不足,本发明的目的是提供一种螺旋纳米碳纤维表面修饰纳米SiO2粒子的方法,以简化现有制备技术中繁琐、复杂的碳纤维前处理工艺过程,减少有机试剂的使用,提高工艺可控性,在螺旋纳米碳纤维表面原位生长粒径可控、分布均匀的球形纳米SiO2粒子。
为实现上述目的,本发明采用如下技术方案:
一种螺旋纳米碳纤维表面修饰纳米SiO2粒子的方法,包括如下步骤:
S1:将螺旋纳米碳纤维在真空条件下于800~1000℃保温2~4h;
S2:将S1中热处理后的螺旋纳米碳纤维分散于乙醇中,超声震荡分散均匀后,再加入蒸馏水和氨水,再次超声震荡分散均匀,此液记为A液;
S3:将正硅酸己酯分散于乙醇中,超声震荡分散均匀,此液记为B液;
S4:将A液于80~100℃冷凝回流后,与B液混合,混合液于70~100℃冷凝回流2~4h;
S5:将S4中得到的混合物抽滤,滤渣干燥后,即得到所述的表面修饰纳米SiO2粒子的螺旋纳米碳纤维。
在上述S1中,热处理温度会影响螺旋碳纳米纤维的石墨化程度的效果,温度过高,会提高螺旋碳纳米纤维的石墨化程度,造成纤维脆化,严重时无法保证纤维的螺旋形貌,温度过低,处理时间短,达不到预期的处理效果,处理时间长,则会造成能源浪费。处理时,可将螺旋纳米碳纤维置于石墨坩埚中保温,为防螺旋纳米碳纤维被加热炉的真空泵抽真空时抽走,可用石墨盖子密封石墨坩埚。
在上述S2中,两次超声震荡时间可为10~20min,乙醇作为分散介质,按照常规要求加入适量即可。
在上述S3中,采用乙醇作为溶剂,相较于丙醇、异丙醇等溶剂,对制备的二氧化硅粒子的效果更好,粒径和形貌更容易控制。超声分散时间可为20~30min,正硅酸己酯与乙醇的体积比可为1:30~150。
在上述S4中,A、B混合液于70~100℃冷凝回流,接枝效果较好,二氧化硅粒子的粒径大小分布均匀、可控,温度过低或过高,都会导致接枝不上,时间过短接枝效果不稳定。A液可预先冷凝回流10~30min,A液若不预先加热,而是混合了B液再加热,那么正硅酸已酯会在升温过程的低温阶段快速水解缩聚生成大粒径的二氧化硅(在30℃左右即可反应生成二氧化硅),而难以接枝在螺旋纳米碳纤维上。
作为优选,S3中螺旋碳纳米纤维与正硅酸己酯的质量体积比为1g:2~6mL。正硅酸己酯过少,接枝效果不明显,正硅酸已酯过多,接上的二氧化硅粒子粒径较大、较多,用量可进一步优选为1g:4mL。
作为优选,S2中加入的蒸馏水、氨水的体积与正硅酸己酯的体积比为1:2~4:0.05~0.2。蒸馏水的作用是与正硅酸已酯发生水解反应提供羟基,氨水是正硅酸己酯水解的催化剂。蒸馏水的量增加可促进正硅酸乙酯的水解使接枝的二氧化硅粒径增大,含量继续增加,硅酸单体浓度开始降低,缩合速率也下降,所接枝SiO2粒径也随之减小;氨水含量增加,可加速整个反应过程的速率,从而使接枝的二氧化硅粒径增大。
作为优选,S5中滤渣于80~100℃真空干燥。干燥时间可为4~8h。
相比现有技术,本发明具有如下有益效果:
1、本发明技术摒弃了在碳纤维表面进行官能团接枝改性的传统工艺,而是利用正硅酸已酯水解后,硅羟基中的氢原子与螺旋纳米碳纤维高温处理后含离域π键的碳原子发生静电吸附作用,提供“原位生长点”来生长纳米SiO2粒子。
2、本发明在对螺旋纳米碳纤维进行前处理的工艺过程中,采用“真空热处理”工艺替代现有技术中的“官能团接枝改性”工艺,克服了现有技术中前处理工艺过于繁琐、复杂的问题,提高了实验的可控性和制备效率。
3、本发明提供的螺旋纳米碳纤维表面修饰纳米SiO2粒子的方法,能在螺旋纳米碳纤维表面均匀地原位生长纳米SiO2粒子,粒径可控,其粒径约为20~100nm。
4、本发明方法工艺简单,操作方便,大幅减少了有机试剂的使用,制备成本低,环保效益显著,适合工业化生产。
附图说明
图1为实施例1制备的表面修饰纳米SiO2粒子的螺旋纳米碳纤维的TEM形貌图;
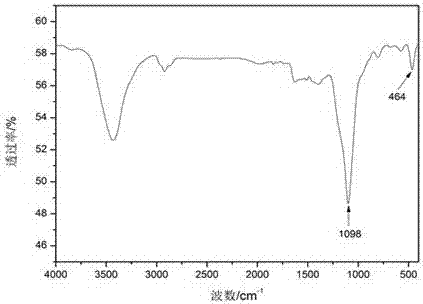
图2为表面修饰纳米SiO2粒子的螺旋纳米碳纤维的红外光谱图。
具体实施方式
下面结合具体实施例对本发明作进一步说明。
本发明实施例中使用的螺旋纳米碳纤维无特殊要求,可选择普通市售商品,使用的石墨坩埚与石墨盖子为螺纹连接。
实施例1:
本实施例中,螺旋纳米碳纤维表面修饰纳米SiO2粒子的方法,包括如下步骤:
S1:将10g的螺旋纳米碳纤维放置在石墨坩埚中,用石墨盖子密封置于真空碳管炉中;抽真空至10-3Pa,从室温升温至1000℃,然后热处理保温4h,待冷却至室温后取出;
S2:称取S1步骤所得的0.1g螺旋纳米碳纤维,并分散于150ml乙醇中,超声震荡10min;再加入4ml蒸馏水和12ml氨水与其混合,再次超声震荡10min,此液记为A液;
S3:将0.4ml正硅酸已酯分散于40ml乙醇中,超声震荡25min,此液记为B液;
S4:将A液置于油浴锅中,在80℃条件下冷凝回流搅拌20min后,再倒入B液,于80℃继续冷凝回流搅拌4h;
S5:将S4所得混合物加适量蒸馏水减压抽滤2次;最后置于真空干燥箱中于100℃干燥4h,得到表面经纳米SiO2粒子修饰的螺旋纳米碳纤维,纳米SiO2的粒径约为30~60nm。
本发明制备的纳米SiO2粒子表面修饰螺旋纳米碳纤维的原理在于:在高温热处理的过程中,利用高温热处理的热活化为螺旋纳米碳纤维的部分碳原子进行重排及结构转变提供能量,促成螺旋纳米碳纤维的局部原子由乱层结构向石墨晶体结构的有序转化,形成少量的离域π键;正硅酸已酯(Si(OC2H5)4)的乙氧基(—OC2H5)水解替换为羟基(—OH),形成羟基化物Si(OH)x(1≤x≤4),由于羟基中的氢原子几乎呈质子状态,可与含离域π键的碳原子发生静电作用,吸附在螺旋纳米碳纤维表面,羟基化物Si(OH)x(1≤x≤4)便可以此为“原位生长点”,脱水缩聚生成纳米SiO2粒子。
图1是实施例1制备的表面修饰纳米SiO2粒子的螺旋纳米碳纤维的TEM形貌图,由图1可知,SiO2均匀地在螺旋纳米碳纤维表面生长,其粒径范围在30~60nm之间。
图2是实施例1表面修饰纳米SiO2粒子的螺旋纳米碳纤维的红外光谱图,由图2可知,在1098cm-1处的Si-O-Si伸缩振动吸收峰最强,464cm-1处Si-O-Si弯曲振动峰次之,表明样品中有大量的桥氧键存在,进一步佐证了螺旋纳米碳纤维表面原位生长的纳米球形粒子为SiO2。
实施例2:
本实施例中,螺旋纳米碳纤维表面修饰纳米SiO2粒子的方法,包括如下步骤:
S1:将10g的螺旋纳米碳纤维放置在石墨坩埚中,用石墨盖子密封置于真空碳管炉中;抽真空至10-3Pa,从室温升温至900℃,然后热处理保温3h,待冷却至室温后取出;
S2:称取S1步骤所得的0.15g螺旋纳米碳纤维,并分散于150ml乙醇中,超声震荡15min,再加入5ml蒸馏水与12ml氨水与其混合,再次超声震荡15min,此液记为A液;
S3:将0.6ml正硅酸已酯分散于60ml乙醇中,超声震荡20min,此液记为B液;
S4:将A液置于油浴锅中,在90℃条件下冷凝回流搅拌15min后倒入B液,继续90℃冷凝回流搅拌3h;
S5:将S4所得混合物加适量蒸馏水减压抽滤3次,置于真空干燥箱中90℃干燥6h,得到表面经纳米SiO2粒子修饰的螺旋纳米碳纤维,纳米SiO2的粒径约为50~80nm。
本实施例制备的螺旋纳米碳纤维TEM图和红外光谱图的测试结果与实施例1结果相似。
实施例3:
本实施例中,螺旋纳米碳纤维表面修饰纳米SiO2粒子的方法,包括如下步骤:
S1:将10g的螺旋纳米碳纤维放置在石墨坩埚中,用石墨盖子密封置于真空碳管炉中;抽真空至10-3Pa,从室温升温至800℃,然后热处理保温2h,待冷却至室温后取出;
S2:称取S1步骤所得的0.1g螺旋纳米碳纤维,并分散于150ml乙醇中,超声震荡15min,再加入5ml蒸馏水与12ml氨水与其混合,再次超声震荡15min,此液记为A液;
S3:将0.4ml正硅酸已酯分散于40ml乙醇中,超声震荡30min,此液记为B液;
S4:将A液置于油浴锅中,在100℃条件下冷凝回流搅拌10min后倒入B液,继续100℃冷凝回流搅拌2h;
S5:将S4所得混合物加适量蒸馏水减压抽滤4次,置于真空干燥箱中80℃干燥8h,得到表面经纳米SiO2粒子修饰的螺旋纳米碳纤维,纳米SiO2的粒径约为20~50nm。
本实施例制备的螺旋纳米碳纤维TEM图和红外光谱图的测试结果与实施例1结果相似。
应用试验
首先,按GB/T 3780.18-2007中的国家标准配方工艺在符合GB 6038规定的混炼设备下进行炼胶,炼胶步骤符合GB/T 3780.18-2007中的混炼程序(开炼机法)。
其中标准配方中的补强剂使用25g的N330炭黑,制得补强橡胶的标准配方试样,在实施例1、2、3中,参照补强橡胶的标准配方试样制作方法,仅将1g表面修饰纳米SiO2粒子的螺旋纳米碳纤维替换10g的N330炭黑,其余配方、制作工艺不变。
对由实施例1~3得到的表面修饰纳米SiO2粒子的螺旋纳米碳纤维取代部分炭黑炼成的胶片和由标准配方工艺得到的胶片的力学性能进行检测,测试5次,去掉最大值和最小值,取平均值,结果如下:
通过以上结果可以看出,由实施例1~3得到的表面修饰纳米SiO2粒子的螺旋纳米碳纤维取代部分炭黑炼成的胶片的力学性能中的拉伸强度、断裂伸长率,都比由标准配方得到胶片的力学性能要高,其中以断裂伸长率最为显著。由此可见,在本发明中表面修饰纳米SiO2粒子的螺旋纳米碳纤维确实具有很好的补强效果,比单一炭黑补强剂的补强效果要好。
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种碳纤维改性纳米绿色环保橡胶及其制备方法与流程
- 一种含纳米碳纤维的高强高弹PVC‑NBR复合发泡板及其制备方法与流程
- 一种用于肿瘤治疗的多孔纳米碳纤维基载药光热试剂及其制备方法与流程
- 一种柔性ZnO纳米晶复合碳纤维及氧化石墨烯葡萄糖探测器及其制备方法与流程
- 一种掺碳纤维纳米片3D打印用改性ABS材料及其制备方法与流程
- 一种有机胺‑TiO2纳米线/碳纤维多尺度增强体的制备方法与流程
- 一种掺碳纤维纳米片3D打印用改性ABS材料及其制备方法与流程
- 碳纤维及塑胶表面用有色纳米单涂UV涂料及其制备方法与流程
- 一种掺碳纤维纳米片及氮化铝纳米颗粒的3D打印用改性ABS材料及其制备方法与流程
- 基于纳米级脱水的污泥深度调理压榨设备的制作方法