一种呋喃生物基聚醚酯共聚物的制备方法、新型呋喃生物基聚醚酯共聚物与流程
本发明属于呋喃基聚醚酯合成技术领域,涉及一种聚醚酯共聚物的制备方法、聚醚酯共聚物,尤其涉及一种呋喃生物基聚醚酯共聚物的制备方法、新型呋喃生物基聚醚酯共聚物。
背景技术
2,5-呋喃二甲酸(fdca),化学式为c6h4o5,结构式为
目前,随着聚酯产品广泛的应用,带动了聚酯原料工业的高速发展。其中,开发用于取代石油基原料的生物基聚酯单体已成为当前聚酯领域研究的热点之一。芳香族聚酯,聚对苯二甲酸乙二醇酯(pet)作为一种重要的热塑性聚酯,具有优良的耐热性、耐化学药品性以及回收率高等优点,广泛地用于包装领域。目前,生产pet的原料之一乙二醇已经可以利用生物质原料进行制备,如现有技术已经成功地以生物基乙二醇为原料制备出了可完全回收的生物基pet饮料瓶。但是,用于生产pet的另一原料对苯二甲酸(pta)是从石油基工业原料对二甲苯(px)的催化氧化制备而来的,致使所得到的pet塑料产品中仅含有30%的植物基成分。虽然,近些年来大力研究的生物基聚酯原料单体丁二酸具有部分替代石油基二酸的潜能,但由于它不能提供像对苯二甲酸一样的刚性芳香苯环结构,极大程度地限制了相应聚酯产品的性能。因此,如何从生物质中获得具有刚性环结构的聚酯原料二元酸是聚酯原料研发领域的一个重要发展方向。
近些年的研究发现,2,5-呋喃二甲酸(fdca)作为一种用于替代pta的理想的聚酯原料研发全生物基聚酯的重要原料,其不仅具有类似的刚性芳香环结构,而且含碳数目,芳香性弱于苯环,更易于降解,更重要的是2,5-呋喃二甲酸是一种可以由生物质制备而来的生物基单体。因而,2,5-呋喃二甲酸相应的聚酯材料,聚2,5呋喃二甲酸乙二醇酯(pef)越来越多地受到了科研人员和企业研发部门的关注。如cn102453242a,cn104072954a,wo2015137804等均报道了pef均聚酯的制备方法及相应高分子产品的热力学性能。目前,除制备pef均聚酯外,通过引入其它的单体与fdca和乙二醇进行共聚,也是开发、拓展呋喃生物基聚酯的种类的一种重要方法,主要是通过引入其它的单体与fdca和乙二醇进行共聚,如cn102432847报道了fdca、乙二醇与对苯二甲酸单体的共聚酯等。
但是这些新引入的共聚单体往往存在成本较高,或是非生物基单体,如对苯二甲酸,这就使得全生物基聚酯产品失去了100%生物基材料的属性,环保意义大大降低。
因此,如何能够得到一种新型的全生物基呋喃生物基聚醚酯高分子材料,不仅具有100%生物基材料的属性,而且能够避免由于加入单体所带来的操作与附加成本等问题,已成为领域内诸多具有前瞻性的研究人员广为关注的焦点之一。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种聚醚酯共聚物的制备方法以及聚醚酯共聚物,特别是一种具有新型结构的呋喃全生物基聚醚酯共聚物的制备方法,本发明提供的制备过程反应过程平稳,易于控制,是一种经济环保,适合规模化工业生产的制备方法;而且制备的呋喃生物基聚醚酯共聚物产品结构中具有高含量甘醇链段,具有较好的热力学性能,而且产品色泽较好。
本发明提供了一种聚醚酯共聚物的制备方法,包括以下步骤:
1`)在保护性气氛和酯化催化剂的条件下,将2,5-呋喃二甲酸、乙二醇和经过酯化反应后,再经过预缩聚反应后,进行缩聚反应,在缩聚反应过程中加入金属配合物催化剂后,得到聚醚酯共聚物。
优选的,所述聚醚酯共聚物中,甘醇链段的摩尔数占所述聚醚酯共聚物的摩尔数的百分比含量为10%~40%;
所述金属配合物的通式为lnx3;
所述金属配合物的配体包括三氟甲磺酸基、五氟乙磺酸基、七氟异丙烷磺酸基、九氟丁烷磺酸基和三氟甲烷磺酰亚胺基中的一种或多种。
优选的,所述金属配合物的金属元素包括稀土元素、锡、铋、锌、铜、碱金属和碱土金属中的一种或多种;
所述酯化催化剂包括氧化亚锡、辛酸亚锡、氯化亚锡、溴化亚锡、碘化亚锡、乙酸亚锡、草酸亚锡、硫酸亚锡和氢氧化亚锡中的一种或多种;
所述2,5-呋喃二甲酸与乙二醇的摩尔比为1:(2~8);
所述金属配合物催化剂的摩尔数占所述2,5-呋喃二甲酸的摩尔数的比值为0.5‰~4‰;
所述酯化催化剂的摩尔数占所述2,5-呋喃二甲酸的摩尔数的比值为1‰~4‰。
优选的,所述酯化反应的温度在170~210℃;所述酯化反应的时间为1~4h;所述酯化反应的压力为1~3atm;
所述预缩聚反应为减压反应;所述预缩聚反应的时间为10~60min;所述预缩聚反应的温度为170~210℃。
优选的,所述预缩聚反应过程的压力为所述酯化反应的压力减压至所述缩聚反应的压力;
所述缩聚反应的压力为20~50pa;所述缩聚反应的温度为180~250℃;所述缩聚反应的时间为2~12h;
所述金属配合物催化剂的加入时间为缩聚反应起始的0~3小时之间。
本发明提供了一种聚醚酯共聚物,具有式(i)所示的结构;
其中,n为10~200;m为0~200;
p选自1、2、3、4和5中的一个或多个。
优选的,所述聚醚酯共聚物中,n/(n+m)为0.1~0.4;
所述聚醚酯共聚物的数均分子量为20000~70000。
优选的,所述聚醚酯共聚物由2,5-呋喃二甲酸和二元醇单体聚合后得到;
所述二元醇单体包括甘醇,或甘醇与乙二醇的混合物;
所述甘醇包括二甘醇、三甘醇、四甘醇和五甘醇中的一种或多种;
所述聚醚酯共聚物中,n/(n+m)小于等于1。
优选的,所述聚醚酯共聚物由以下方法制备得到:
1)在保护性气氛和金属配合物催化剂条件下,将2,5-呋喃二甲酸和乙二醇经过酯化反应后,再经过预缩聚反应和缩聚反应后,得到聚醚酯共聚物。
优选的,所述聚醚酯共聚物中,n/(n+m)为0.3~0.7;
所述酯化反应的温度在170~210℃;所述酯化反应的时间为1~4h;所述酯化反应的压力为1~3atm;
所述预缩聚反应为减压反应;所述预缩聚反应的时间为10~60min;所述预缩聚反应的温度为170~210℃;
所述缩聚反应的压力为20~50pa;所述缩聚反应的温度为180~250℃;所述缩聚反应的时间为2~12h。
本发明提供了一种聚醚酯共聚物的制备方法,包括以下步骤,在保护性气氛和酯化催化剂的条件下,将2,5-呋喃二甲酸、乙二醇和经过酯化反应后,再经过预缩聚反应后,进行缩聚反应,在缩聚反应过程中加入金属配合物催化剂后,得到聚醚酯共聚物。与现有技术相比,本发明针对现有的聚2,5呋喃二甲酸乙二醇酯,特别是共聚酯,需要通过引入其它的单体与fdca和乙二醇进行共聚,而这些新引入的共聚单体往往存在成本较高,或是非生物基单体,进而就使得全生物基聚酯产品失去了100%生物基材料的属性,环保意义大大降低的缺陷。
本发明创造性的提供了一种具有新型结构的呋喃全生物基聚醚酯共聚物的制备方法,本发明在金属配合物催化剂的催化作用下,利用含强吸电子配体的金属催化剂在较高温度下能够催化醚化反应的特点,采用简单的合成手段,利用fdca与生物基乙二醇直接进行缩聚反应,在较低的温度下,较短的时间内,高效地制备高粘度的具有新型结构的呋喃全生物基聚醚酯共聚物,其分子中含有含量可控的低聚甘醇链段,而且反应过程平稳,易于控制,条件温和,是一种经济环保,适合规模化工业生产的制备方法。本发明提供的呋喃生物基聚醚酯共聚物产品结构中具有高含量甘醇链段,具有较好的热力学性能,而且产品色泽较好;同时该呋喃基聚醚酯共聚物还能够通过普通的低聚甘醇、fdca和乙二醇进行共聚,只是低聚甘醇(二甘醇、三甘醇等)为非生物基单体,使得由低聚甘醇与fdca和乙二醇进行共聚所制备的产品失去了100%生物基材料的属性。
本发明提供的具有新型结构的呋喃全生物基聚醚酯共聚物的制备方法,既可以得到一种新型的全生物基高分子材料,又可以解决制备过程中由于加入低聚甘醇所带来的操作与附加成本等问题。该新型结构的呋喃生物基聚醚酯产品,推动实现对呋喃类可再生资源的开发和利用,以及对环境友好的新型生物基聚酯的制备,逐步摆脱对石油资源的依赖,对解决我国聚合物工业面临的资源短缺和环境污染等问题,实现我国高分子材料产业的可持续发展具有重要的推动作用及应用价值。
实验结果表明,本发明制备的新型生物基聚醚酯的玻璃化转变温度可以控制在30~85℃,5%热降解温度在280~400,拉伸强度在10mpa~85mpa,断裂伸长率在4%~400%。
附图说明
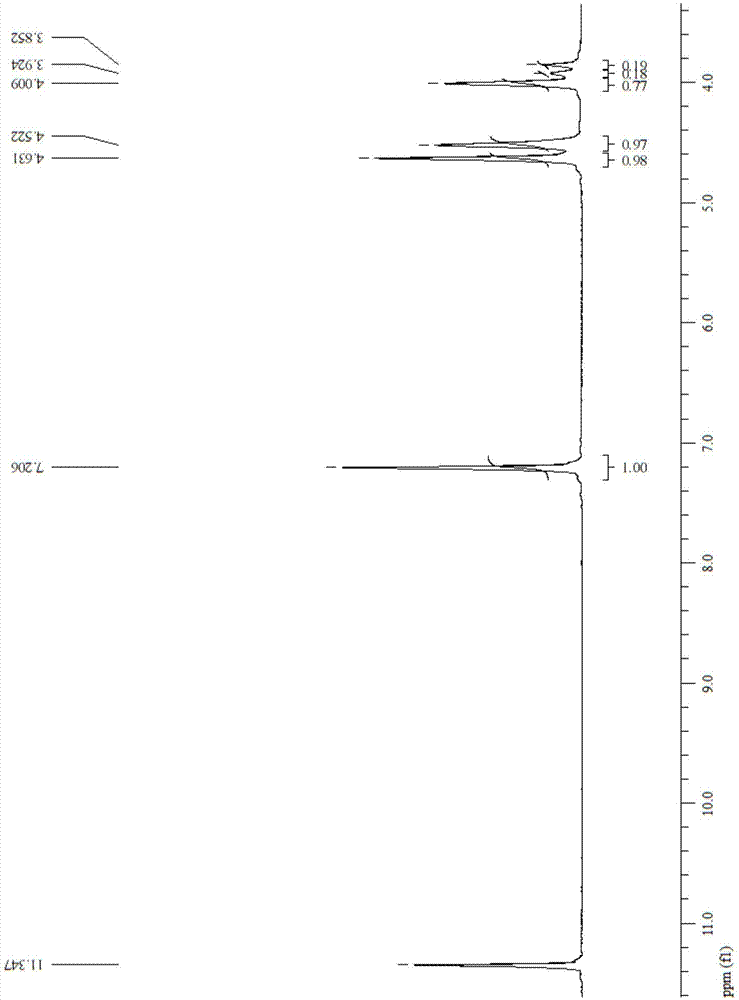
图1为本发明实施例1制备的提供的呋喃基聚醚酯共聚酯的核磁共振氢谱图。
具体实施方式
为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为了进一步说明本发明的特征和优点,而不是对发明权利要求的限制。
本发明所有原料,对其来源没有特别限制,在市场上购买的或按照本领域技术人员熟知的常规方法制备的即可。
本发明所有原料,对其纯度没有特别限制,本发明优选采用分析纯或呋喃基聚醚酯合成技术领域的常规纯度。
本发明所有名词表达和简称均属于本领域常规的名词表达和简称,每个名词表达和简称在其相关应用领域内均是清楚明确的,本领域技术人员根据该名词表达和简称,能够清楚准确唯一的进行理解。
本发明提供了一种聚醚酯共聚物的制备方法,包括以下步骤:
1`)在保护性气氛和酯化催化剂的条件下,将2,5-呋喃二甲酸、乙二醇和经过酯化反应后,再经过预缩聚反应后,进行缩聚反应,在缩聚反应过程中加入金属配合物催化剂后,得到聚醚酯共聚物。
本发明对所述聚醚酯共聚物的结构没有特别限制,以本领域技术人员熟知的聚醚酯共聚物的结构即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述聚醚酯共聚物中,甘醇链段的摩尔数占所述聚醚酯共聚物的摩尔数的百分比含量优选为10%~40%,更优选为15%~35%,更优选为20%~30%。本发明所述2,5-呋喃二甲酸优选包括生物基2,5-呋喃二甲酸。
本发明对所述保护性气氛没有特别限制,以本领域技术人员熟知的保护性气氛即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述保护性气氛优选包括氮气和/或惰性气体,更优选为氮气或氩气。
本发明对所述酯化催化剂没有特别限制,以本领域技术人员熟知的用于此类化合物常规的酯化催化剂即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述酯化催化剂优选包括氧化亚锡、辛酸亚锡、氯化亚锡、溴化亚锡、碘化亚锡、乙酸亚锡、草酸亚锡、硫酸亚锡和氢氧化亚锡中的一种或多种,更优选为氧化亚锡、辛酸亚锡、氯化亚锡、溴化亚锡、碘化亚锡、乙酸亚锡、草酸亚锡、硫酸亚锡或氢氧化亚锡。本发明对所述酯化催化剂的用量没有特别限制,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述酯化催化剂的摩尔数占所述2,5-呋喃二甲酸的摩尔数的比值优选为0.5‰~4‰,更优选为1‰~3.5‰,更优选为1.5‰~3‰,更优选为2‰~2‰。
本发明对所述乙二醇的用量没有特别限制,以本领域技术人员熟知的用于此类反应的常规用量即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述2,5-呋喃二甲酸与乙二醇的摩尔比优选为1:(2~8),更优选为1:(3~7),更优选为1:(4~6)。
本发明对所述酯化反应的条件没有特别限制,以本领域技术人员熟知的常规酯化反应的条件即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述酯化反应的温度优选为170~210℃,更优选为175~205℃,更优选为180~200℃,更优选为185~195℃。本发明所述酯化反应的时间优选为1~4h,更优选为1.5~3.5h,更优选为2~3h。本发明所述酯化反应的压力可以为常压或微正压,具体优选为1~3atm(大气压,可以为0.1mpa),更优选为1.2~2.8atm,更优选为1.5~2.5atm。
本发明进行酯化反应后,酯化产物无需分离,直接进行预缩聚反应。本发明对所述预缩聚反应的条件和步骤没有特别限制,以本领域技术人员熟知的常规预缩聚反应的条件和步骤即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述预缩聚反应的温度优选为170~210℃,更优选为175~205℃,更优选为180~200℃,更优选为185~195℃。本发明所述预缩聚反应的时间优选为10~60min,更优选为20~50min,更优选为30~40min。
本发明所述预缩聚反应,优选为减压反应,具体反应方式可以为减压蒸馏反应,同时还能除去体系中过量的二元醇。本发明所述预缩聚反应的压力优选为一个变化值,其变化范围具体可以为所述酯化反应的压力减压至所述缩聚反应的压力。
本发明经过上述预缩聚反应后,最后在低真空条件下进行缩聚反应。本发明对所述缩聚反应的条件和步骤没有特别限制,以本领域技术人员熟知的常规缩聚反应的条件和步骤即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述缩聚反应的压力优选为20~50pa,更优选为25~45pa,更优选为30~40pa。所述缩聚反应的温度优选为180~250℃,更优选为190~240℃,更优选为200~230℃,更优选为210~220℃。本发明所述缩聚反应的时间优选为2~12h,更优选为4~10h,更优选为6~8h。
本发明特别在缩聚反应中加入金属配合物催化剂,其中所述缩聚反应中可以为缩聚反应开始或缩聚反应过程中。本发明对所述加入的具体时间没有特别限制,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述金属配合物催化剂的加入时间优选为缩聚反应开始时的0~3小时之间,即缩聚反应的0~3小时,也可以为0.5~2.5小时,也可以为1~2小时。
本发明对所述金属配合物催化剂的具体选择没有特别限制,以本领域技术人员熟知的常规的金属配合物催化剂即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述金属配合物的通式可以为lnx3。本发明所述金属配合物催化剂的配体优选包括三氟甲磺酸基、五氟乙磺酸基、七氟异丙烷磺酸基、九氟丁烷磺酸基和三氟甲烷磺酰亚胺基中的一种或多种,更优选为三氟甲磺酸基、五氟乙磺酸基、七氟异丙烷磺酸基、九氟丁烷磺酸基或三氟甲烷磺酰亚胺基。本发明所述金属配合物的金属元素优选包括稀土元素、锡、铋、锌、铜、碱金属和碱土金属中的一种或多种,更优选为稀土元素、锡、铋、锌、铜、碱金属或碱土金属。
本发明对所述稀土元素的具体选择没有特别限制,以本领域技术人员熟知的常用的稀土元素即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述稀土元素优选为无辐射的稀土元素,具体可以为镧la、铈ce、镨pr、铕eu、钕nd、钐sm、钆ga、镝dy、钬ho、铒er、铥tm、镱yb、钇y和钪sc中的一种或多种。
本发明对所述金属配合物催化剂的用量没有特别限制,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述金属配合物催化剂的摩尔数占所述2,5-呋喃二甲酸的摩尔数的比值优选为0.5‰~4‰,更优选为1‰~3.5‰,更优选为1.5‰~3‰,更优选为2‰~2‰。
本发明为进一步保证最终产品的性能,完整和细化制备过程,上述制备方法具体可以为:
采用熔融缩聚法,使2,5-呋喃二甲酸与二元醇(乙二醇)经过酯化、预聚合和聚合三步过程,直接制备酯化与醚化结构比例在10%~40%的可控的新型呋喃全生物基聚醚酯共聚物。先在常规酯化催化剂(pef酯化催化剂)的作用下,2,5-呋喃二甲酸先与生物基二元醇直接酯化生成2,5-呋喃二甲酸二醇酯;而后,酯化产物无需分离,继续利用减压蒸馏进行预缩聚并除去反应体系中过量的二元醇;而后,可以在不同时间内,加入催化剂lnx3,并在低真空条件下进行缩聚反应制备高分子量聚醚酯产品。
本发明上述制备过程具体可以如反应式(2)所示:
本发明上述步骤提供了一种新型呋喃全生物基聚醚酯共聚物的制备方法,按照该方法制备的新型全生物基聚醚酯的结构链段中,主要包含部分聚呋喃二甲酸乙二醇酯单元(pef)及部分甘醇酯链段,其中,甘醇链段包括二甘醇及三甘醇,也可以还有部分四甘醇和/或五甘醇。所制备的聚醚酯结构中的甘醇链段占聚合物总体摩尔百分含量为10%~40%,即n/(n+m)为0.1~0.4,本发明所述n优选为10~200,更优选为30~180,更优选为50~150,更优选为80~120。本发明所述m优选为10~200,更优选为30~180,更优选为50~150,更优选为80~120。在本发明中,所述m和n均优选为摩尔数。
本发明采用金属配合物催化剂催化二醇与呋喃二甲酸直接酯化聚合,在较低的温度下,较短的时间内,高效地制备高粘度的新型的100%生物基呋喃聚醚酯,反应过程平稳,易于控制,产品色泽较好,所制备的产品结构中具有较高的可控含量的甘醇链段。而本发明还能够通过调整二元醇与呋喃二甲酸的投料比例,以及反应过程中的酯化时间、温度,缩聚时间、温度等条件可以有效的调控聚醚酯产品中甘醇链段的比例。更进一步的,本发明特别优选了稀土金属配合物催化剂,其低度或无毒,使得所制备的新型高分子材料更加符合绿色环保的理念,适合规模化工业生产的制备方法。
本发明提供了一种聚醚酯共聚物,具有式(i)所示的结构;
其中,n为10~200;m为0~200;
p选自1、2、3和4中的一个或多个。
本发明对所述聚醚酯共聚物中的选择、组合和优选范围,优选与前述聚醚酯共聚物的制备方法中的选择、组合和优选范围中能够对应,在此不再一一赘述。
本发明对所述具有式(i)所示结构的聚醚酯共聚物没有其他特别限制,本领域技术人员能够理解,该聚醚酯共聚物可以为该结构式,也可以含有该结构的片段,本发明不做特别限制。本发明所述具有式(i)所示结构的聚醚酯共聚物,其结构的核心在于,是一种含有聚2,5-呋喃二甲酸乙二醇酯与聚呋喃二甲酸与多醇酯共聚合的共聚物,其链段中含有2,5-呋喃二甲酸乙二醇酯片段,也含有2,5-呋喃二甲酸多甘醇酯片段;其简称可以为写为:ppegf,名称可以为聚(2,5-呋喃二甲酸乙二醇酯-co-2,5-呋喃二甲酸多甘醇酯)
本发明式(i)所示结构中,n所对应的部分,是2,5-呋喃二甲酸多甘醇酯片段,因而对所述n的选择范围没有特别限制,以本领域技术人员熟知的常规范围即可,本领域技术人员可以根据实际应用情况、质量控制以及产品要求进行选择和调整,本发明所述n优选为10~200,更优选为30~180,更优选为50~150,更优选为80~120。本发明式(i)所示结构中,m所对应的部分,是2,5-呋喃二甲酸二元醇酯片段,因而对所述m的选择范围没有特别限制,以本领域技术人员熟知的常规范围即可,本领域技术人员可以根据实际应用情况、质量控制以及产品要求进行选择和调整,本发明所述m优选为0~200,更优选为10~200,更优选为30~180,更优选为50~150,更优选为80~120。在本发明中,所述m和n均优选为摩尔数。
本发明式(i)所示结构中,p所对应的部分,是多甘醇的具体元数,本领域技术人员可以根据实际应用情况、质量控制以及产品要求进行选择和调整,本发明所述多甘醇可以包括为二甘醇、三甘醇、四甘醇和五甘醇中的一种或多种,即p优选自1、2、3和4中的一个或多个,更优选为1、2、3或4,更优选为1、1和2、1和2和3、1和2和3和4,或者,1和2和3和4和5。
本发明对所述n与m的具体比例没有特别限制,以本领域技术人员熟知的常规摩尔比即可,本领域技术人员可以根据实际应用情况、质量控制以及产品要求进行选择和调整,本发明所述聚醚酯共聚物中,n/(n+m)优选小于等于1,可以等于1,即m为0,也可以小于等于0.8,也可以小于等于0.6,也可以小于等于0.4,或者小于等于0.2。本发明所述聚醚酯共聚物中,依据制备方法的不同,n/(n+m)可以为0.3~0.7,也可以为0.35~0.65,也可以为0.4~0.6,也可以为0.45~0.55。本发明所述聚醚酯共聚物中,依据制备方法的不同,n/(n+m)可以为0.1~0.4,也可以为0.15~0.35,也可以为0.2~0.3。
本发明对所述聚醚酯共聚物的其他参数没有特别限制,以本领域技术人员熟知的聚醚酯共聚物的常规参数即可,本领域技术人员可以根据实际应用情况、质量控制以及产品要求进行选择和调整,本发明所述聚醚酯共聚物的数均分子量优选为20000~70000,更优选为30000~60000,更优选为40000~50000。
本发明对所述聚醚酯共聚物的制备方法没有特别限制,以本领域技术人员熟知的常规此类共聚物的制备方法即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述聚醚酯共聚物优选由2,5-呋喃二甲酸和醇单体聚合后得到。
本发明所述醇单体特别包括甘醇,或甘醇与乙二醇的混合物。
本发明对所述甘醇的具体选择没有特别限制,本领域技术人员可以根据实际应用情况、质量控制以及产品要求进行选择和调整,本发明所述甘醇优选包括二甘醇、三甘醇、四甘醇和五甘醇中的一种或多种,更优选为二甘醇、三甘醇、四甘醇或五甘醇。
本发明对所述聚合的具体方式和步骤没有特别限制,以本领域技术人员熟知的常规的聚合和方式即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整。
本发明为进一步提高聚醚酯共聚物产品的可控性,开发呋喃全生物基聚醚酯共聚物,完善和细化制备过程,为工业化应用提高更完整的技术方案,本发明提供了另外两种独立的呋喃基聚醚酯共聚物的制备方法。
本发明还提供了一种通常聚醚酯共聚物的制备方法,包括以下步骤:
1)在保护性气氛和金属配合物催化剂条件下,将2,5-呋喃二甲酸和乙二醇经过酯化反应后,再经过预缩聚反应和缩聚反应后,得到聚醚酯共聚物。
本发明对所述聚醚酯共聚物的制备方法中的聚醚酯共聚物的选择、组合和优选范围,优选与前述聚醚酯共聚物中的选择、组合和优选范围能够对应,在此不再一一赘述。
本发明对所述聚醚酯共聚物的结构没有特别限制,以本领域技术人员熟知的聚醚酯共聚物的结构即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述聚醚酯共聚物中,甘醇链段的摩尔数占所述聚醚酯共聚物的摩尔数的百分比含量优选为30%~70%,即更优选为35%~65%,更优选为40%~60%,更优选为45%~55%。本发明所述2,5-呋喃二甲酸优选包括生物基2,5-呋喃二甲酸。
本发明对所述保护性气氛没有特别限制,以本领域技术人员熟知的保护性气氛即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述保护性气氛优选包括氮气和/或惰性气体,更优选为氮气或氩气。
本发明对所述金属配合物催化剂的具体选择没有特别限制,以本领域技术人员熟知的常规的金属配合物催化剂即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述金属配合物的通式可以为lnx3。本发明所述金属配合物催化剂的配体优选包括三氟甲磺酸基、五氟乙磺酸基、七氟异丙烷磺酸基、九氟丁烷磺酸基和三氟甲烷磺酰亚胺基中的一种或多种,更优选为三氟甲磺酸基、五氟乙磺酸基、七氟异丙烷磺酸基、九氟丁烷磺酸基或三氟甲烷磺酰亚胺基。本发明所述金属配合物的金属元素优选包括稀土元素、锡、铋、锌、铜、碱金属和碱土金属中的一种或多种,更优选为稀土元素、锡、铋、锌、铜、碱金属或碱土金属。
本发明对所述稀土元素的具体选择没有特别限制,以本领域技术人员熟知的常用的稀土元素即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述稀土元素优选为无辐射的稀土元素,具体可以为镧la、铈ce、镨pr、铕eu、钕nd、钐sm、钆ga、镝dy、钬ho、铒er、铥tm、镱yb、钇y和钪sc中的一种或多种。
本发明对所述金属配合物催化剂的用量没有特别限制,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述金属配合物催化剂的摩尔数占所述2,5-呋喃二甲酸的摩尔数的比值优选为0.5‰~4‰,更优选为1‰~3.5‰,更优选为1.5‰~3‰,更优选为2‰~2.5‰。
本发明对所述乙二醇的用量没有特别限制,以本领域技术人员熟知的用于此类反应的常规用量即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述2,5-呋喃二甲酸与乙二醇的摩尔比优选为1:(2~8),更优选为1:(3~7),更优选为1:(4~6)。
本发明对所述酯化反应的条件没有特别限制,以本领域技术人员熟知的常规酯化反应的条件即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述酯化反应的温度优选为170~210℃,更优选为175~205℃,更优选为180~200℃,更优选为185~195℃。本发明所述酯化反应的时间优选为1~4h,更优选为1.5~3.5h,更优选为2~3h。本发明所述酯化反应的压力可以为常压或微正压,具体优选为1~3atm(大气压,可以为0.1mpa),更优选为1.2~2.8atm,更优选为1.5~2.5atm。
本发明进行酯化反应后,酯化产物无需分离,直接进行预缩聚反应。本发明对所述预缩聚反应的条件和步骤没有特别限制,以本领域技术人员熟知的常规预缩聚反应的条件和步骤即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述预缩聚反应的温度优选为170~210℃,更优选为175~205℃,更优选为180~200℃,更优选为185~195℃。本发明所述预缩聚反应的时间优选为10~60min,更优选为20~50min,更优选为30~40min。
本发明所述预缩聚反应,优选为减压反应,具体反应方式可以为减压蒸馏反应,同时还能除去体系中过量的二元醇。本发明所述预缩聚反应的压力优选为一个变化值,其变化范围具体可以为所述酯化反应的压力减压至所述缩聚反应的压力。
本发明经过上述预缩聚反应后,最后在低真空条件下进行缩聚反应。本发明对所述缩聚反应的条件和步骤没有特别限制,以本领域技术人员熟知的常规缩聚反应的条件和步骤即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述缩聚反应的压力优选为20~50pa,更优选为25~45pa,更优选为30~40pa。所述缩聚反应的温度优选为180~250℃,更优选为190~240℃,更优选为200~230℃,更优选为210~220℃。本发明所述缩聚反应的时间优选为2~12h,更优选为4~10h,更优选为6~8h。
本发明为进一步保证最终产品的性能,完整和细化制备过程,上述制备方法具体可以为:
采用熔融缩聚法,使2,5-呋喃二甲酸与二元醇(乙二醇)经过酯化、预聚合和聚合三步过程,直接制备酯化与醚化结构比例在30%~70%可控的新型呋喃全生物基聚醚酯共聚物。在金属配合物催化剂作用下,2,5-呋喃二甲酸先与生物基二元醇直接酯化生成2,5-呋喃二甲酸二醇酯;而后,酯化产物无需分离,继续利用减压蒸馏进行预缩聚并除去反应体系中过量的二元醇;最终,在低真空条件下进行缩聚反应制备高分子量聚醚酯产品。
本发明上述制备过程具体可以如反应式(1)所示:
本发明上述步骤提供了一种新型呋喃全生物基聚醚酯共聚物的制备方法,按照该方法制备的新型全生物基聚醚酯的结构链段中,主要包含部分聚呋喃二甲酸乙二醇酯单元(pef)及部分甘醇酯链段,其中,甘醇链段包括二甘醇及三甘醇,也可以还有部分四甘醇和/或五甘醇。所制备的聚醚酯结构中的甘醇链段占聚合物总体摩尔百分含量为30%~70%,即n/(n+m)为0.3~0.7,本发明所述n优选为10~200,更优选为30~180,更优选为50~150,更优选为80~120。本发明所述m优选为10~200,更优选为30~180,更优选为50~150,更优选为80~120。在本发明中,所述m和n均优选为摩尔数。
本发明采用金属配合物催化剂催化二醇与呋喃二甲酸直接酯化聚合,在较低的温度下,较短的时间内,高效地制备高粘度的新型的100%生物基呋喃聚醚酯,反应过程平稳,易于控制,产品色泽较好,所制备的产品结构中具有高含量甘醇链段。而本发明还能够通过调整二元醇与呋喃二甲酸的投料比例,以及反应过程中的酯化时间、温度,缩聚时间、温度等条件可以有效的调控聚醚酯产品中甘醇链段的比例。更进一步的,本发明特别优选了稀土金属配合物催化剂,其低度或无毒,使得所制备的新型高分子材料更加符合绿色环保的理念,适合规模化工业生产的制备方法。
本发明还提供了一种通常聚醚酯共聚物的制备方法,包括以下步骤:
1``)在保护性气氛和酯化催化剂的条件下,将2,5-呋喃二甲酸、醇单体经过酯化反应后,再经过预缩聚反应和缩聚反应,得到聚醚酯共聚物。
本发明对所述聚醚酯共聚物的制备方法中的聚醚酯共聚物的选择、组合和优选范围,优选与前述聚醚酯共聚物中的选择、组合和优选范围能够对应,在此不再一一赘述。
本发明对所述聚醚酯共聚物的结构没有特别限制,以本领域技术人员熟知的聚醚酯共聚物的结构即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述聚醚酯共聚物中,甘醇链段的摩尔数占所述聚醚酯共聚物的摩尔数的百分比含量优选为0.1%~100%,更优选为10%~90%,更优选为20%~80%,更优选为30%~70%,更优选为40%~60%。本发明所述2,5-呋喃二甲酸优选包括生物基2,5-呋喃二甲酸。
本发明对所述保护性气氛没有特别限制,以本领域技术人员熟知的保护性气氛即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述保护性气氛优选包括氮气和/或惰性气体,更优选为氮气或氩气。
本发明对所述酯化催化剂没有特别限制,以本领域技术人员熟知的用于此类化合物常规的酯化催化剂即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述酯化催化剂优选包括氧化亚锡、辛酸亚锡、氯化亚锡、溴化亚锡、碘化亚锡、乙酸亚锡、草酸亚锡、硫酸亚锡和氢氧化亚锡中的一种或多种,更优选为氧化亚锡、辛酸亚锡、氯化亚锡、溴化亚锡、碘化亚锡、乙酸亚锡、草酸亚锡、硫酸亚锡或氢氧化亚锡。本发明对所述酯化催化剂的用量没有特别限制,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述酯化催化剂的摩尔数占所述2,5-呋喃二甲酸的摩尔数的比值优选为0.5‰~4‰,更优选为1‰~3.5‰,更优选为1.5‰~3‰,更优选为2‰~2‰。
本发明对所述醇单体的选择没有特别限制,以本领域技术人员熟知的常规醇单体即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述醇单体优选包括甘醇,或甘醇与乙二醇的混合物。本发明所述甘醇优选包括二甘醇、三甘醇、四甘醇和五甘醇中的一种或多种,更优选为二甘醇或三甘醇。
在本发明中,当醇单体为甘醇时,所述聚醚酯共聚物的式(i)结构中,n=1;所述醇单体为甘醇与乙二醇的混合物时,所述聚醚酯共聚物的式(i)结构中,n/(n+m)小于1。在本发明中,当采用上述制备方式时,由于使用了甘醇,在甘醇不能生物基化(通过生物原料完全制备)之前,其最终产品可以认为是非全生物基聚醚酯共聚物。
本发明对所述醇单体的用量没有特别限制,以本领域技术人员熟知的用于此类反应的常规用量即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述2,5-呋喃二甲酸与醇单体的摩尔比优选为1:(2~8),更优选为1:(3~7),更优选为1:(4~6)。
本发明对所述酯化反应的条件没有特别限制,以本领域技术人员熟知的常规酯化反应的条件即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述酯化反应的温度优选为170~210℃,更优选为175~205℃,更优选为180~200℃,更优选为185~195℃。本发明所述酯化反应的时间优选为1~4h,更优选为1.5~3.5h,更优选为2~3h。本发明所述酯化反应的压力可以为常压或微正压,具体优选为1~3atm(大气压,可以为0.1mpa),更优选为1.2~2.8atm,更优选为1.5~2.5atm。
本发明进行酯化反应后,酯化产物无需分离,直接进行预缩聚反应。本发明对所述预缩聚反应的条件和步骤没有特别限制,以本领域技术人员熟知的常规预缩聚反应的条件和步骤即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述预缩聚反应的温度优选为170~210℃,更优选为175~205℃,更优选为180~200℃,更优选为185~195℃。本发明所述预缩聚反应的时间优选为10~60min,更优选为20~50min,更优选为30~40min。
本发明所述预缩聚反应,优选为减压反应,具体反应方式可以为减压蒸馏反应,同时还能除去体系中过量的二元醇。本发明所述预缩聚反应的压力优选为一个变化值,其变化范围具体可以为所述酯化反应的压力减压至所述缩聚反应的压力。
本发明经过上述预缩聚反应后,最后在低真空条件下进行缩聚反应。本发明对所述缩聚反应的条件和步骤没有特别限制,以本领域技术人员熟知的常规缩聚反应的条件和步骤即可,本领域技术人员可以根据实际生产情况、质量控制以及产品要求进行选择和调整,本发明所述缩聚反应的压力优选为20~50pa,更优选为25~45pa,更优选为30~40pa。所述缩聚反应的温度优选为180~250℃,更优选为190~240℃,更优选为200~230℃,更优选为210~220℃。本发明所述缩聚反应的时间优选为2~12h,更优选为4~10h,更优选为6~8h。
本发明为进一步保证最终产品的性能,完整和细化制备过程,上述制备方法具体可以为:
采用熔融缩聚法,使2,5-呋喃二甲酸与醇单体经过酯化、预聚合和聚合三步过程,直接制备酯化与醚化结构比例在0.1%~100%的可控的新型呋喃聚醚酯共聚物。在常规酯化催化剂(pef酯化催化剂)的作用下,2,5-呋喃二甲酸先与醇单体直接酯化生成2,5-呋喃二甲酸二醇酯和2,5-呋喃二甲酸二甘醇酯;而后,酯化产物无需分离,继续利用减压蒸馏进行预缩聚并除去反应体系中过量的醇单体;而后,在低真空条件下进行缩聚反应制备高分子量聚醚酯产品。
本发明上述步骤提供了一种新型呋喃聚醚酯共聚物的制备方法,按照该方法制备的新型全生物基聚醚酯的结构链段中,主要包含部分聚呋喃二甲酸乙二醇酯单元(pef),及部分或全部甘醇酯链段,其中,甘醇链段包括二甘醇及三甘醇,也可以还有部分四甘醇和/或五甘醇。所制备的聚醚酯结构中的甘醇链段占聚合物总体摩尔百分含量为0.1%~100%,即n/(n+m)可以小于等于1,本发明所述n优选为10~200,更优选为30~180,更优选为50~150,更优选为80~120。本发明所述m优选为0~200,更优选为10~200,更优选为30~180,更优选为50~150,更优选为80~120。在本发明中,所述m和n均优选为摩尔数。
本发明采用酯化催化剂催化醇单体与呋喃二甲酸直接酯化聚合,在较低的温度下,较短的时间内,高效地制备高粘度的新型的呋喃聚醚酯共聚物,反应过程平稳,易于控制,产品色泽较好,所制备的产品结构中具有较高的可控含量的甘醇链段。而本发明还能够通过调整醇单体与呋喃二甲酸的投料比例,以及反应过程中的酯化时间、温度,缩聚时间、温度等条件可以有效的调控聚醚酯产品中甘醇链段的比例。
本发明上述步骤提供了一种聚醚酯共聚物及其多种制备方法,该聚醚酯共聚物是一种具有新型结构的呋喃全生物基聚醚酯共聚物,本发明提供的呋喃生物基聚醚酯共聚物产品结构中具有高含量甘醇链段,具有较好的热力学性能,而且产品色泽较好。而且本发明提供的呋喃基聚醚酯共聚物不仅能够通过普通的低聚甘醇、fdca和乙二醇进行共聚,更考虑到低聚甘醇(二甘醇、三甘醇等)为非生物基单体,使得由低聚甘醇与fdca和乙二醇进行共聚所制备的产品失去了100%生物基材料的属性。本发明更在金属配合物催化剂,利用含强吸电子配体的金属催化剂在较高温度下能够催化醚化反应的特点,特别是稀土金属配合物催化剂的催化作用下,采用简单的合成手段,利用fdca与生物基乙二醇直接进行缩聚反应,在较低的温度下,较短的时间内,高效地制备高粘度的具有新型结构的呋喃全生物基聚醚酯共聚物,其分子中含有含量可控的低聚甘醇链段,而且反应过程平稳,易于控制,条件温和,是一种经济环保,适合规模化工业生产的制备方法。
本发明提供的具有新型结构的呋喃全生物基聚醚酯共聚物制备方法及新型结构的呋喃全生物基聚醚酯共聚物,既可以得到一种新型的全生物基高分子材料,又可以解决制备过程中由于加入低聚甘醇所带来的操作与附加成本等问题。该新型结构的呋喃生物基聚醚酯产品,推动实现对呋喃类可再生资源的开发和利用,以及对环境友好的新型生物基聚酯的制备,逐步摆脱对石油资源的依赖,对解决我国聚合物工业面临的资源短缺和环境污染等问题,实现我国高分子材料产业的可持续发展具有重要的推动作用及应用价值。
实验结果表明,本发明制备的新型生物基聚醚酯的玻璃化转变温度可以控制在30~85℃,5%热降解温度在280~400,拉伸强度在10mpa~85mpa,断裂伸长率在4%~400%。
为了进一步理解本发明,下面结合实施例对本发明提供的一种聚醚酯共聚物的制备方法、聚醚酯共聚物进行说明,但是应当理解,这些实施例是在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制,本发明的保护范围也不限于下述的实施例。
实施例1
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲磺酸钐87毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的聚(2,5-呋喃二甲酸乙二醇酯-co-2,5-呋喃二甲酸二甘醇酯-co-2,5-呋喃二甲酸三甘醇酯),记为ppegf。
对本发明实施例1制备的呋喃基聚醚酯共聚物进行表征,以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析。
参见图1,图1为本发明实施例1制备的提供的呋喃基聚醚酯共聚酯的核磁共振氢谱图。由图1可知,该ppegf共聚酯具有式(ii)所示的结构,其中乙二醇链段与甘醇链段的比例为1:1,结构中的甘醇链段包括二甘醇与三甘醇。
将上述制备的ppegf共聚酯,溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.91dl/g。
对本发明实施例制备的呋喃基聚醚酯共聚物进行性能检测。
参见表1,表1为本发明实施例制备的典型结构呋喃基聚醚酯的性能。
表1
实施例2
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为1:1,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为1.01dl/g。
实施例3
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲磺酸钕85毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为1:1,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.83dl/g。
实施例4
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲磺酸镧85毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为1:1,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.96dl/g。
实施例5
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲磺酸铈85毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为1:1,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.69dl/g。
实施例6
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲磺酸铥85毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为1:1,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.94dl/g。
实施例7
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入五氟乙磺酸基钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得深棕色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为1:1,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为1.00dl/g。
实施例8
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入七氟异丙基钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得深棕色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为1:1,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.95dl/g。
实施例9
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酰亚胺基钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得深棕色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为1:1,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为1.01dl/g。
实施例10
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸铋92毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得深棕色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为1:1,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为1.05dl/g。
实施例11
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪58毫克(占二元羧酸单体总量的0.12%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为1:1,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.96dl/g。
实施例12
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇12.0克(0.2mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化2小时,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为65:35,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.92dl/g。
实施例13
于反应器中,分别加入2,5-呋喃二甲酸15.6克(1mol)、乙二醇15.0克(0.25mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为60:40,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.82dl/g。
实施例14
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇24.0克(0.4mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为45:55,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.98dl/g。
实施例15
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇30.0克(0.5mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为43:57,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.71dl/g。
实施例16
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在170℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为60:40,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.90dl/g。
实施例17
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在190℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为44:56,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.98dl/g。
实施例18
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在200℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为37:63,结构中的甘醇链段包括二甘醇与三甘醇、四甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.81dl/g。
实施例19
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在210℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为30:70,结构中的甘醇链段包括二甘醇、三甘醇、四甘醇、五甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.65dl/g。
实施例20
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化60分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为68:32,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.81dl/g。
实施例21
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化90分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为55:45,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.86dl/g。
实施例22
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化150分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为45:55,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.75dl/g。
实施例23
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化210分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为42:58,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.57dl/g。
实施例24
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至180℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为42:58,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.45dl/g。
实施例25
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至190℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为44:56,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.50dl/g。
实施例26
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至200℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为47:53,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.85dl/g。
实施例27
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至220℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为56:44,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为1.08dl/g。
实施例28
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应1小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为46:54,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.65dl/g。
实施例29
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应2小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为46:54,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.70dl/g。
实施例30
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应2.5小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为49:51,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.80dl/g。
实施例31
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入三氟甲烷磺酸钪72毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在180℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,升温至210℃,25分钟内将压力将至20~50pa,熔融缩聚反应3小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为50:50,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.87dl/g。
实施例32
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入草酸亚锡24毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在210℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,在210℃,25分钟内将压力将至20~50pa,熔融缩聚反应0.5小时,加入三氟甲磺酸钪72毫克,继续缩聚3.5小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为78:22,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.70dl/g。
实施例33
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇18.0克(0.3mol),加入草酸亚锡24毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在210℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,在210℃,25分钟内将压力将至20~50pa,熔融缩聚反应3小时,加入三氟甲磺酸钪72毫克,继续缩聚1小时,制得黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与甘醇链段的比例为90:10,结构中的甘醇链段包括二甘醇与三甘醇。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.78dl/g。
实施例34
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇6.0克(0.1mol),二甘醇10.6克(0.1mol),加入草酸亚锡24毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在210℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,在230℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时。制得结构含有二甘醇和乙二醇链段的黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与二甘醇链段的比例为1:1.4。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.63dl/g。
实施例35
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、乙二醇6.0克(0.1mol),三甘醇15克(0.1mol),加入草酸亚锡24毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在210℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,在230℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时。制得结构含有三甘醇和乙二醇链段的黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与三甘醇链段的比例为1:3。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.65dl/g。
实施例36
于反应器中,分别加入2,5-呋喃二甲酸15.6克(0.1mol)、二甘醇10.6克(0.1mol),三甘醇15克(0.1mol),加入草酸亚锡24毫克(占二元羧酸单体总量的0.15%),氮气氛围下,在210℃条件下酯化105分钟,得到酯化产物;酯化反应结束后,在230℃,25分钟内将压力将至20~50pa,熔融缩聚反应4小时。制得结构含有三甘醇和二甘醇链段的黄色的ppegf共聚物。
以氘代三氟醋酸为溶剂对所述ppegf共聚酯进行核磁共振分析,所述ppegf共聚酯结构中的乙二醇链段与三甘醇链段的比例为1:2。
将所述ppegf共聚酯溶解于25℃质量比为1:1的苯酚和四氯乙烷混合溶剂中测定其特性粘度,其特性粘度为0.60dl/g。
以上对本发明提供的一种呋喃生物基聚醚酯共聚物的制备方法、新型呋喃生物基聚醚酯共聚物进行了详细的介绍,本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想,包括最佳方式,并且也使得本领域的任何技术人员都能够实践本发明,包括制造和使用任何装置或系统,和实施任何结合的方法。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。本发明专利保护的范围通过权利要求来限定,并可包括本领域技术人员能够想到的其他实施例。如果这些其他实施例具有不是不同于权利要求文字表述的结构要素,或者如果它们包括与权利要求的文字表述无实质差异的等同结构要素,那么这些其他实施例也应包含在权利要求的范围内。
- 还没有人留言评论。精彩留言会获得点赞!