一种含异苯并二氢吡喃-1-酮骨架的化合物的制备方法与流程
本发明涉及含异色满酮骨架化合物的制备方法,具体涉及一种含异苯并二氢吡喃-1-酮骨架的化合物的制备方法。
技术背景
异苯并二氢吡喃-1-酮骨架结构因具有抗真菌,抗细胞毒素,抗疟疾,抗过敏等活性而成为许多药物中间体的基本骨架组成,[参见:(a)chem.pharm.bull.1996,44,1440–1447;(b)j.nat.prod.2006,69,612–615.]其中绝大多数含异苯并二氢吡喃-1-酮骨架结构的化合物是从各种天然产物(如植物、昆虫和微生物)中分离而得的,此外也有一些关于制备含异苯并二氢吡喃-1-酮骨架结构化合物的报道,目前常用于制备含异苯并二氢吡喃-1-酮骨架结构化合物的合成方法包括:分子间的环合反应[参见:(a)j.org.chem.2016,81,5752–5758(b)med.chem.2008,16,9383-9391.]、分子内的环合反应[参见:org.lett.2006,8,5517–5520.]、重排反应[参见:tetrahedronlett.1998,54,8737–8744.]等。但是这些方法中往往涉及到有腐蚀性的强酸、强碱的使用或者是反应步骤较为繁琐、反应时间较长,因而使其应用范围受到了一定的局限性。
技术实现要素:
本发明的目的是提供一种含异苯并二氢吡喃-1-酮骨架的化合物的制备方法,该合成方法具有原料廉价易得、方法简单、一步合成等优点。
本发明为实现上述目的而采取的技术方案为:
一种含异苯并二氢吡喃-1-酮骨架的化合物的制备方法,包括如下步骤:
(1)按摩尔比1-3:1:0.1:1:0.2-3.1:0.1将取代基取代的1,1-二取代烯烃、取代基取代的邻卤苯甲酸甲酯、醋酸钯、碳酸银、配体和对甲苯磺酸一水合物加入反应器中,加入溶剂将反应物溶解,在室温下混合均匀,在130℃下反应12~48小时;
(2)反应完成后将反应器冷却至室温,加入乙酸乙酯溶解,依次用饱和氯化铵和饱和氯化钠水溶液洗涤,有机相经无水硫酸钠干燥后,过滤,在旋转蒸发仪上旋去溶剂;
(3)将旋去溶剂后的剩余物用硅胶柱层析分离纯化,收集目标产物,旋蒸除去溶剂,油泵抽干;
所述的溶剂为等体积的1,1,1,3,3,3-六氟-2-丙醇和邻二甲苯的混合物;所述配体为膦配体,也可以为乙酰基保护的氨基酸和dmso的混合配体。
所述的取代基取代的1,1-二取代烯烃、取代基取代的邻卤苯甲酸甲酯、醋酸钯、碳酸银、配体和对甲苯磺酸一水合的物摩尔比优选为2:1:0.1:2:0.2-3.1:0.1。
所述的取代基取代的1,1二取代烯烃为α-甲基苯乙烯、1,1-二苯乙烯、1-甲基-4-(1-甲基乙烯基)苯、对氯甲基苯乙烯、4-硝基-α-甲基苯乙烯、4-(丙-1-烯-2-基)苯甲酸甲酯等;所述的取代基取代的邻卤苯甲酸甲酯为:邻碘苯甲酸甲酯、2-溴-5-甲氧基苯甲酸甲酯、2-溴-4-甲基苯甲酸甲酯等。
所述膦配体优选为:三(4-三氟甲苯基)膦、三(4-氟苯基)膦、三(五氟苯基)膦、三(环己基)膦、三(2-甲氧基苯基)膦、三(2-呋喃基)膦或四氟硼酸三叔丁基膦。
所述的乙酰基保护的氨基酸为:n-乙酰-l-半胱氨酸、n-乙酰-l-苯丙氨酸、n-乙酰-l-异亮氨酸、n-乙酰-l-亮氨酸、n-乙酰-l-酪氨酸、n-乙酰-l-蛋氨酸、n-乙酰-l-缬氨酸或n-乙酰甘氨酸。
本发明的技术路线是取代基取代的1,1-二取代烯烃与取代基取代的邻卤苯甲酸甲酯的偶联合环反应,其化学方程式为:
其中,r1为甲基、甲氧基;r2为氢原子、甲基、氯原子、硝基、甲酯基;r3为甲基、苯基。
本发明采用核磁共振氢谱(1hnmr)、碳谱(13cnmr)以及高分辨质谱证实了含异苯并二氢吡喃-1-酮骨架化合物的结构。检测所用仪器为:avanceiiihd600mhz型核磁共振仪,其中氘代氯仿为内标(氢谱,氘代氯仿:δ7.26ppm).(碳谱,氘代氯仿:δ77ppm)。thermoscientificqexactive型高分辨质谱仪。
与现有合成方法相比,本发明的优点具体体现为:
(1)本发明所采用的方法是交叉偶联反应,官能团适用范围及官能团的容忍性都较广,包括:含卤素、烷基、苯基、烷氧基、酯基、噻吩等取代基取代的底物;
(2)本发明所用合成路线为直接交叉偶联反应,与传统合成反应相比较,反应步骤简单,提高了合成总产率,降低了总成本;
(3)本发明中的合成未使用任何导向基团,无需额外步骤引入或脱除导向基团;
(4)本发明所用合成路线为快速高效得到含有环内酯骨架结构的复杂天然产物提供了新的途径。
附图说明
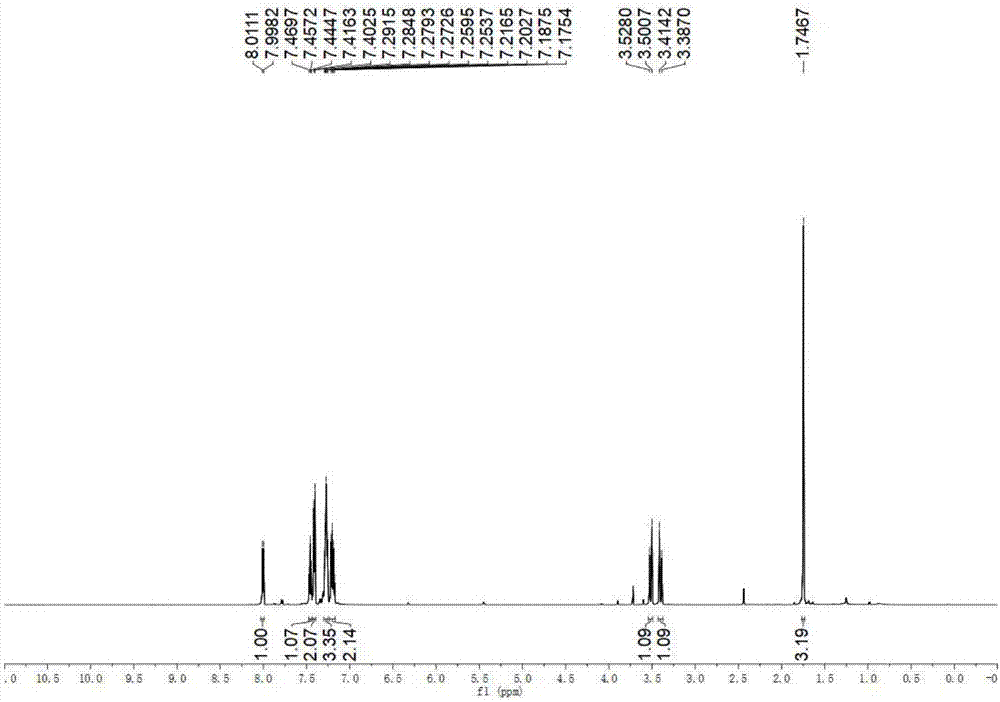
图1为3-甲基-3-苯基异苯并二氢吡喃-1-酮的氢谱;
图2为3-甲基-3-苯基异苯并二氢吡喃-1-酮的碳谱;
图3为3,3-二苯基异色满-1-酮的氢谱;
图4为3,3-二苯基异色满-1-酮的碳谱;
图5为3-甲基-3-(对甲苯基)异苯并二氢吡喃-1-酮的氢谱;
图6为3-甲基-3-(对甲苯基)异苯并二氢吡喃-1-酮的碳谱;
图7为3-甲基-3-(4-氯苯基)异苯并二氢吡喃-1-酮的氢谱;
图8为3-甲基-3-(4-氯苯基)异苯并二氢吡喃-1-酮的碳谱;
图9为3-甲基-3-(4-硝基苯基)异苯并二氢吡喃-1-酮的氢谱;
图10为3-甲基-3-(4-硝基苯基)异苯并二氢吡喃-1-酮的碳谱;
图11为4-(3-甲基-1-氧代异色满-3-基)苯甲酸甲酯的氢谱;
图12为4-(3-甲基-1-氧代异色满-3-基)苯甲酸甲酯的碳谱。
图13为7-甲氧基-3-甲基-3-苯基异苯并二氢吡喃-1-酮的氢谱;
图14为7-甲氧基-3-甲基-3-苯基异苯并二氢吡喃-1-酮的碳谱;
图15为3,6-二甲基-3-苯基异苯并二氢吡喃-1-酮的氢谱;
图16为3,6-二甲基-3-苯基异苯并二氢吡喃-1-酮的碳谱;
具体实施方式
下面结合具体实施例对本发明作进一步详细描述,将有助于对本发明的理解,但并不是以此来限制本发明的保护范围。
实施例1:3-甲基-3-苯基异苯并二氢吡喃-1-酮的合成
(1)将邻碘苯甲酸甲酯(0.037ml,0.25mmol),α-甲基苯乙烯(0.065ml,0.5mmol),醋酸钯(0.0056g,0.025mmol),三(2-甲氧基苯基)膦(0.0176g,0.05mmol),碳酸银(0.1379g,0.5mmol),对甲苯磺酸一水合物(0.0048g,0.025mmol),1,1,1,3,3,3-六氟-2-丙醇(0.5ml),邻二甲苯(0.5ml),在干净干燥的密闭反应管中搅拌均匀后加热到130℃,反应24小时。
(2)反应完成后将反应管冷却至室温,加入30ml乙酸乙酯将反应体系稀释并移入100ml的分液漏斗中,加入20ml饱和氯化铵水溶液,摇晃,静置,移除下层的水相后,再加入20ml饱和食盐水,摇晃,静置,移除下层水相,将有机相用无水硫酸钠干燥,减压移去溶剂,剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到无色油状物,目标产物45.3mg,产率76%。氢谱和碳谱如图1和图2所示,1hnmr(600mhz,cdcl3)δ8.00(d,j=7.7hz,1h),7.46(t,j=7.5hz,1h),7.41(d,j=8.3hz,2h),7.27(dt,j=11.4,3.7hz,3h),7.20(dd,j=16.9,7.8hz,2h),3.51(d,j=16.3hz,1h),3.40(d,j=16.3hz,1h),1.75(s,3h).;13cnmr(151mhz,cdcl3)δ165.11,143.51,137.75,133.79,129.81,128.39,127.55,127.40,127.34,125.07,124.54,83.50,38.96,30.03.hrms(esi+):计算值c16h15o2[m+h]+:239.1067,实测值239.1065。
实施例2:3,3-二苯基异色满-1-酮的合成
(1)将邻碘苯甲酸甲酯(0.037ml,0.25mmol),1,1-二苯乙烯(0.088ml,0.50mmol),醋酸钯(0.0056g,0.025mmol),三(4-三氟甲苯基)膦(0.0233g,0.05mmol),碳酸银(0.1379g,0.5mmol),对甲苯磺酸一水合物(0.0048g,0.025mmol),1,1,1,3,3,3-六氟-2-丙醇(0.5ml),邻二甲苯(0.5ml),在干净干燥的密闭反应管中搅拌均匀后加热到130℃,反应48小时。
(2)反应完成后将反应管冷却至室温,加入30ml乙酸乙酯将反应体系稀释并移入100ml的分液漏斗中,加入20ml饱和氯化铵水溶液,摇晃,静置,移除下层的水相后,再加入20ml饱和食盐水,摇晃,静置,移除下层水相,将有机相用无水硫酸钠干燥,减压移去溶剂,剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到黄色油状物,目标产物39.1mg,产率52%。氢谱和碳谱如图3和图4所示,1hnmr(600mhz,cdcl3)δ7.98(dd,j=7.7,0.8hz,1h),7.50(td,j=7.5,1.2hz,1h),7.43(dd,j=8.3,0.9hz,4h),7.32(d,j=7.5hz,1h),7.30–7.26(m,5h),7.23–7.19(m,2h),3.82(s,2h).;13cnmr(151mhz,cdcl3)δ165.01,142.96,138.10,134.04,130.12,128.40,127.64,127.53,127.43,126.08,125.61,86.48,39.00.hrms(esi+):计算值c21h17o2[m+h]+:301.1223,实测值301.1224。
实施例3:3-甲基-3-(对甲苯基)异苯并二氢吡喃-1-酮的合成
(1)将邻碘苯甲酸甲酯(0.037ml,0.25mmol),1-甲基-4-(1-甲基乙烯基)苯(0.073ml,0.50mmol),醋酸钯(0.0056g,0.025mmol),三(4-三氟甲苯基)膦(0.0233g,0.05mmol),碳酸银(0.1379g,0.5mmol),对甲苯磺酸一水合物(0.0048g,0.025mmol),1,1,1,3,3,3-六氟-2-丙醇(0.5ml),邻二甲苯(0.5ml),在干净干燥的密闭反应管中搅拌均匀后加热到130℃,反应24小时。
(2)反应完成后将反应管冷却至室温,加入30ml乙酸乙酯将反应体系稀释并移入100ml的分液漏斗中,加入20ml饱和氯化铵水溶液,摇晃,静置,移除下层的水相后,再加入20ml饱和食盐水,摇晃,静置,移除下层水相,将有机相用无水硫酸钠干燥,减压移去溶剂,剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到无色油状物,目标产物59.2mg,产率94%。氢谱和碳谱如图5和图6所示,1hnmr(600mhz,cdcl3)δ7.99(d,j=7.7hz,1h),7.44(t,j=7.5hz,1h),7.30–7.24(m,3h),7.20(d,j=7.6hz,1h),7.06(d,j=8.1hz,2h),3.50(d,j=16.3hz,1h),3.38(d,j=16.3hz,1h),2.24(s,3h),1.73(s,3h).;13cnmr(151mhz,cdcl3)δ165.21,140.55,137.89,137.00,133.74,129.81,129.06,127.54,127.35,125.14,124.51,83.52,38.95,30.21,20.77.hrms(esi+):计算值c17h17o2[m+h]+:253.1223,实测值253.1223。
实施例4:3-(4-氯苯基)-3-甲基异苯并二氢吡喃-1-酮的合成
(1)将邻碘苯甲酸甲酯(0.037ml,0.25mmol),4-氯-α-甲基苯乙烯(0.072ml,0.50mmol),醋酸钯(0.0056g,0.025mmol),三(4-三氟甲苯基)膦(0.0233g,0.05mmol),碳酸银(0.1379g,0.5mmol),对甲苯磺酸一水合物(0.0048g,0.025mmol),1,1,1,3,3,3-六氟-2-丙醇(0.5ml),邻二甲苯(0.5ml),在干净干燥的密闭反应管中搅拌均匀后加热到130℃,反应48小时。
(2)反应完成后将反应管冷却至室温,加入30ml乙酸乙酯将反应体系稀释并移入100ml的分液漏斗中,加入20ml饱和氯化铵水溶液,摇晃,静置,移除下层的水相后,再加入20ml饱和食盐水,摇晃,静置,移除下层水相,将有机相用无水硫酸钠干燥,减压移去溶剂,剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到浅黄色油状物,目标产物61.3mg,产率90%。氢谱和碳谱如图7和图8所示,1hnmr(600mhz,cdcl3)δ8.01(d,j=7.7hz,1h),7.49(t,j=7.5hz,1h),7.35(d,j=7.6hz,2h),7.32(t,j=7.6hz,1h),7.25(d,j=8.4hz,2h),7.22(d,j=7.5hz,1h),3.48(d,j=16.3hz,1h),3.40(d,j=16.3hz,1h),1.74(s,3h).;13cnmr(151mhz,cdcl3)δ164.91,142.22,137.51,134.02,133.39,130.09,128.71,127.71,127.59,126.18,125.04,83.14,39.01,30.07.hrms(esi+):计算值c17h14clo2[m+h]+:273.0677,实测值273.0676。
实施例5:3-甲基-3-(4-硝基苯基)异苯并二氢吡喃-1-酮的合成
(1)将邻碘苯甲酸甲酯(0.037ml,0.25mmol),4-硝基-α-甲基苯乙烯(0.08159g,0.5mmol),醋酸钯(0.0056g,0.025mmol),n-乙酰-l-半胱氨酸(0.00716g,0.05mmol),dmso(0.05ml),碳酸银(0.1379g,0.5mmol),对甲苯磺酸一水合物(0.0048g,0.025mmol),1,1,1,3,3,3-六氟-2-丙醇(0.5ml),邻二甲苯(0.5ml),在干净干燥的密闭反应管中搅拌均匀后加热到130℃,反应48小时。
(2)反应完成后将反应管冷却至室温,加入30ml乙酸乙酯将反应体系稀释并移入100ml的分液漏斗中,加入20ml饱和氯化铵水溶液,摇晃,静置,移除下层的水相后,再加入20ml饱和食盐水,摇晃,静置,移除下层水相,将有机相用无水硫酸钠干燥,减压移去溶剂,剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-5:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到浅黄色油状物,目标产物51.7mg,产率73%。氢谱和碳谱如图9和图10所示,1hnmr(600mhz,cdcl3)δ8.15(d,j=8.1hz,2h),8.02(d,j=7.7hz,1h),7.62(d,j=8.0hz,2h),7.51(t,j=7.5hz,1h),7.33(t,j=7.6hz,1h),7.24(d,j=7.6hz,1h),3.52(d,j=16.4hz,1h),3.47(d,j=16.4hz,1h),1.78(s,3h).;13cnmr(151mhz,cdcl3)δ164.44,150.83,147.20,136.95,134.30,130.20,127.99,127.64,125.77,124.74,123.85,83.00,38.92,29.66.hrms(esi+):计算值c17h13no4[m+h]+:284.0917,实测值284.0918。
实施例6:4-(3-甲基-1-氧代异色满-3-基)苯甲酸甲酯的合成
(1)将邻碘苯甲酸甲酯(0.037ml,0.25mmol),4-(丙-1-烯-2-基)苯甲酸甲酯(0.08811g,0.5mmol),醋酸钯(0.0056g,0.025mmol),n-乙酰-l-半胱氨酸(0.00716g,0.05mmol),dmso(0.05ml),碳酸银(0.1379g,0.5mmol),对甲苯磺酸一水合物(0.0048g,0.025mmol),1,1,1,3,3,3-六氟-2-丙醇(0.5ml),邻二甲苯(0.5ml),在干净干燥的密闭反应管中搅拌均匀后加热到130℃,反应48小时。
(2)反应完成后将反应管冷却至室温,加入30ml乙酸乙酯将反应体系稀释并移入100ml的分液漏斗中,加入20ml饱和氯化铵水溶液,摇晃,静置,移除下层的水相后,再加入20ml饱和食盐水,摇晃,静置,移除下层水相,将有机相用无水硫酸钠干燥,减压移去溶剂,剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-5:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到浅黄色油状物,目标产物50.1mg,产率68%。氢谱和碳谱如图11和图12所示,1hnmr(600mhz,cdcl3)δ8.01(d,j=7.6hz,1h),7.95(d,j=7.6hz,2h),7.48(dd,j=15.5,7.7hz,3h),7.30(t,j=7.4hz,1h),7.21(d,j=7.4hz,1h),3.87(s,3h),3.51(d,j=16.3hz,1h),3.44(d,j=16.3hz,1h),1.76(s,3h).;13cnmr(151mhz,cdcl3)δ166.49,164.86,148.61,137.38,134.04,130.07,129.86,129.39,127.74,127.59,125.01,124.75,83.37,52.11,39.04,29.83.hrms(esi+):计算值c18h17o4[m+h]+:297.1121,实测值297.1124。
实施例7:7-甲氧基-3-甲基-3-苯基异苯并二氢吡喃-1-酮的合成
(1)将2-溴-5-甲氧基苯甲酸甲酯(0.040ml,0.25mmol),α-甲基苯乙烯(0.065ml,0.50mmol),醋酸钯(0.0056g,0.025mmol),三(4-三氟甲苯基)膦(0.0233g,0.05mmol),碳酸银(0.1379g,0.5mmol),对甲苯磺酸一水合物(0.0048g,0.025mmol),1,1,1,3,3,3-六氟-2-丙醇(0.5ml),邻二甲苯(0.5ml),在干净干燥的密闭反应管中搅拌均匀后加热到130℃,反应12小时。
(2)反应完成后将反应管冷却至室温,加入30ml乙酸乙酯将反应体系稀释并移入100ml的分液漏斗中,加入20ml饱和氯化铵水溶液,摇晃,静置,移除下层的水相后,再加入20ml饱和食盐水,摇晃,静置,移除下层水相,将有机相用无水硫酸钠干燥,减压移去溶剂,剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到黄色油状物,目标产物50.8mg,产率76%。氢谱和碳谱如图13和图14所示,1hnmr(600mhz,cdcl3)δ7.50(d,j=2.5hz,1h),7.40(d,j=8.3hz,2h),7.28(t,j=7.7hz,2h),7.20(t,j=7.3hz,1h),7.12(d,j=8.3hz,1h),7.02(dd,j=8.3,2.6hz,1h),3.77(s,3h),3.46(d,j=16.2hz,1h),3.34(d,j=16.2hz,1h),1.74(s,3h).;13cnmr(151mhz,cdcl3)δ165.34,158.76,143.65,130.06,128.71,128.46,127.40,125.93,124.62,121.65,112.73,83.94,55.42,38.23,30.11.hrms(esi+):计算值c17h17o3[m+h]+:269.1172,实测值269.1172。
实施例8:3,6-二甲基-3-苯基异苯并二氢吡喃-1-酮的合成
(1)将2-溴-4-甲基苯甲酸甲酯(0.0573g,0.25mmol),α-甲基苯乙烯(0.065ml,0.50mmol),醋酸钯(0.0056g,0.025mmol),三(4-三氟甲苯基)膦(0.0233g,0.05mmol),碳酸银(0.1379g,0.5mmol),对甲苯磺酸一水合物(0.0048g,0.025mmol),1,1,1,3,3,3-六氟-2-丙醇(0.5ml),邻二甲苯(0.5ml),在干净干燥的密闭反应管中搅拌均匀后加热到130℃,反应12小时。
(2)反应完成后将反应管冷却至室温,加入30ml乙酸乙酯将反应体系稀释并移入100ml的分液漏斗中,加入20ml饱和氯化铵水溶液,摇晃,静置,移除下层的水相后,再加入20ml饱和食盐水,摇晃,静置,移除下层水相,将有机相用无水硫酸钠干燥,减压移去溶剂,剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到无色油状物,目标产物56.6mg,产率90%。氢谱和碳谱如图15和图16所示,1hnmr(600mhz,cdcl3)δ7.90(d,j=7.8hz,1h),7.41(d,j=7.6hz,2h),7.31–7.26(m,2h),7.20(t,j=7.3hz,1h),7.09(d,j=7.9hz,1h),7.01(s,1h),3.46(d,j=16.3hz,1h),3.36(d,j=16.3hz,1h),2.34(s,3h),1.74(s,3h).;13cnmr(151mhz,cdcl3)δ165.32,144.80,143.76,137.81,129.99,128.45,128.40,128.14,127.36,124.62,122.50,83.42,39.08,30.14,21.69.hrms(esi+):计算值c17h17o2[m+h]+:253.1223,实测值253.1223。
实施例9:3-甲基-3-苯基异苯并二氢吡喃-1-酮的合成
(1)将邻碘苯甲酸甲酯(0.037ml,0.25mmol),α-甲基苯乙烯(0.065ml,0.5mmol),醋酸钯(0.0056g,0.025mmol),n-乙酰-l-亮氨酸(0.00866g,0.05mmol),dmso(0.05ml),碳酸银(0.1379g,0.5mmol),对甲苯磺酸一水合物(0.0048g,0.025mmol),1,1,1,3,3,3-六氟-2-丙醇(0.5ml),邻二甲苯(0.5ml),在干净干燥的密闭反应管中搅拌均匀后加热到130℃,反应24小时。
(2)反应完成后将反应管冷却至室温,加入30ml乙酸乙酯将反应体系稀释并移入100ml的分液漏斗中,加入20ml饱和氯化铵水溶液,摇晃,静置,移除下层的水相后,再加入20ml饱和食盐水,摇晃,静置,移除下层水相,将有机相用无水硫酸钠干燥,减压移去溶剂,剩余物用硅胶柱层析(石油醚/乙酸乙酯=20:1-10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,得到无色油状物,目标产物48.8mg,产率82%。氢谱和碳谱如图1和图2所示,1hnmr(600mhz,cdcl3)δ8.00(d,j=7.7hz,1h),7.46(t,j=7.5hz,1h),7.41(d,j=8.3hz,2h),7.27(dt,j=11.4,3.7hz,3h),7.20(dd,j=16.9,7.8hz,2h),3.51(d,j=16.3hz,1h),3.40(d,j=16.3hz,1h),1.75(s,3h).;13cnmr(151mhz,cdcl3)δ165.11,143.51,137.75,133.79,129.81,128.39,127.55,127.40,127.34,125.07,124.54,83.50,38.96,30.03.hrms(esi+):计算值c16h15o2[m+h]+:239.1067,实测值239.1065。
- 还没有人留言评论。精彩留言会获得点赞!