一种单齿配位噁唑啉配体及其制备方法和应用与流程
本发明属于医药中间体合成领域,具体涉及一种单齿配位噁唑啉配体及其制备方法和应用。
背景技术:
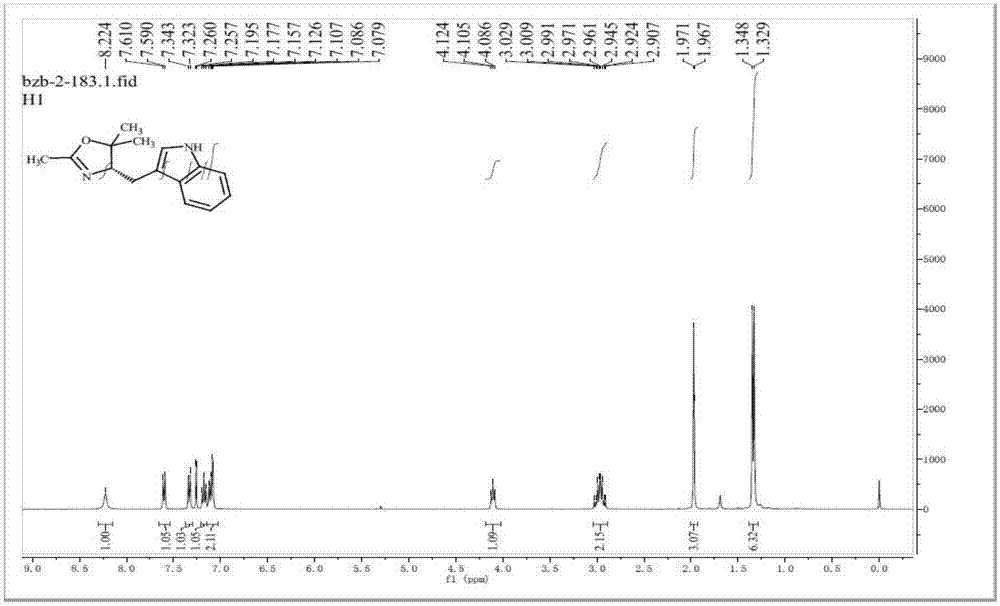
手性化合物在天然化合物中是广泛存在,并且已经发展成为治疗各种疾病的有效药物。而天然化合物对于社会发展的需求是远远不够的,所以需要我们化学家去合成各种各样的手性分子以满足我们的需求。传统的有机合成是无法合成单一手性化合物的,需要复杂的拆分程序才能得到单一的手性化合物。因此不对称合成是得到单一手性化合物的最有效的手段,近年来得到了非常巨大的发展。2001年诺贝尔化学奖授予美国科学家威廉·诺尔斯,巴里·夏普莱斯与日本科学家野依良治,以表彰他们在“手性催化反应”领域所作出的贡献。足以看出不对称催化是一个非常重要而且亟待继续发展的领域。所谓的不对称催化,就是使用一个确定的单一构型的催化剂,以实现对反应底物的选择性控制。而在金属催化中,金属原子本身是不具有手性的,因此一种高效的方法就是使用天然中存在的手性分子合成具有单一手性的手性配体。而手性配体的设计合成是不对称催化中的关键环节,只有合成出各种各样的手性配体,才能实现不对称合成的多样性,从而得到更多有用的手性化合物。众所周知,噁唑啉是一类具有应用非常广泛的手性配体,并且发展出许多知名的配体,例如双噁唑啉配体(box,pybox),双齿的n,n-,p-n,s-n等配体以及唐勇院士发展的三噁唑啉配体等多齿配位的噁唑啉配体,由于多齿配位可以很有效的与金属中心形成高效的手性催化剂,提供有效的手性环境,可以实现各类化学反应的高效的不对称合成。由于其高效的手性环境构筑能力是单齿噁唑啉配体所不能实现的,因此单齿噁唑啉配体的发展一直非常缓慢。但是单齿噁唑啉配体是一类更简单易得的手性配体,但是其应用是非常局限的,几乎没有关于单齿噁唑啉配体的催化反应报道。因此,设计新型单齿噁唑啉配体并将其利用于有效的不对称催化反应中是非常重要的,可以用于合成各类含有手性中心的药物分子以及医药中间体分子,具有非常重要的科研意义与工业应用的前景。技术实现要素:本发明的目的是针对现有技术的不足,提供一种单齿配位噁唑啉配体及其制备方法和应用,本发明单齿配位噁唑啉配体可以首次并有效应用于烯烃的不对称官能团化反应中,且取得了最高90%ee的立体选择性。一种单齿配位噁唑啉配体,所述单齿配位噁唑啉配体的结构通式如下:其中,r1为-ch3;r2为h、苯基、3,5-双三氟甲基苯基、五氟苯基或对甲氧基苯基中的一种;r3为h或-ch3;本发明还提供了配体l1或l2单齿配位噁唑啉配体的制备方法,包括以下步骤:(1)在相应的非极性含卤有机溶剂中,将式i所示的化合物、式ii所示的化合物、有机酸催化剂按照1:1.2-2.0:0.05-0.1的摩尔配比混合均匀,在50-100度下回流反应10-24小时,分离纯化,得到式iii所示的化合物,具体的合成路线如下所示:进一步地,所述步骤(1)中的非极性含卤有机溶剂为1,2-二氯乙烷或二氯甲烷中的一种;所述有机酸催化剂为醋酸,甲酸,丙酸,苯基酸,邻苯基苯基酸中的一种;进一步地,步骤(1)中的反应还可以在氮气保护下进行。本发明还提供了配体l3、l4、l5或l6单齿配位噁唑啉配体的制备方法,包括以下步骤:(1)在相应的非极性含卤有机溶剂中,将式i所示的化合物、式ii所示的化合物、有机酸催化剂按照1:1.2-2.0:0.05-0.1的摩尔配比混合均匀,在50-100度下回流反应10-24小时,分离纯化,得到式iii所示的化合物,具体的合成路线如下所示:(2)在相应的苯系有机溶剂中,将式iii所示的化合物、式iv所示的卤化合物、碘化亚铜、n,n'-二甲基-1,2-环己二胺、磷酸钾按照1:1.1-2.0:0.05-0.2:0.10-0.4:1.2-2.0的摩尔配比,在40-90度下反应6-24小时,得到式v所示的配体,具体的合成路线如下所示:进一步地,所述步骤(1)中的非极性含卤有机溶剂为1,2-二氯乙烷或二氯甲烷中的一种;所述有机酸催化剂为醋酸,甲酸,丙酸,苯甲酸,邻甲基苯甲酸中的一种;所述步骤(2)中的苯系有机溶剂为甲苯、二甲苯或氯苯中的一种。进一步地,步骤(1)或步骤(2)中的反应还可以在氮气保护下进行,并可以取得较空气更好的产率与品质。本发明还提供了单齿配位噁唑啉配体的应用,包括以下步骤:将式vi所示的底物、亲核试剂、钯催化剂、l1-l6所示的配体中的一种,邻苯基苯甲酸(o-pba)和醇系有机溶剂按照1:1.5:0.1:0.3:1.0的摩尔比混合,在室温下搅拌30分钟,随后在60oc的加热块上反应48小时。将反应液冷却至室温,得到式vii所示的化合物,且式vii所示的化合物的ee值为60-90%。进一步地,所述醇系有机溶剂为甲醇、乙醇或异丙醇中的一种;所述钯催化剂为醋酸钯催化剂、二苯甲酸钯催化剂;所述亲核试剂为取代吲哚。进一步地,所述单齿配位噁唑啉配体的应用反应还可以在氩气保护下进行。有益效果本发明单齿配位噁唑啉配体相对于多齿配体,简单易得,制备方法高效,具有其独特的手性控制活性,应用在不对称反应中选择性高。本发明的单齿噁唑啉实现了首例钯催化的双导向的非活化烯烃的不对称氢碳官能团化反应.各种类型的吲哚均可以高效的实现c3-位置的不对称偶联反应,可以以高达90%ee的立体选择性反应得到γ-加成的产物。并且该手性噁唑啉配体可以使用廉价的氨基酸高效制备,合成非常简单,可以高效的应用于高效合成含有手性分子的医药中间体,是单齿噁唑啉配体的重要研究进展。附图说明图1为l1配体的氢谱;图2为l1配体的碳谱;图3为l2配体的氢谱;图4为l2配体的碳谱;图5为l3配体的氢谱;图6为l3配体的碳谱;图7为l4配体的氢谱;图8为l4配体的碳谱;图9为l5配体的氢谱;图10为l5配体的碳谱;图11为l6配体的氢谱;图12为l6配体的碳谱;图13为实施例9-14中式vii所示的化合物的氢谱;图14为实施例9-14中式vii所示的化合物的碳谱;图15为实施例12中式vii所示的不对称产物的hplc的谱图;图16为实施例12中式vii所示的消旋产物的hplc的谱图;图17为实施例15-20中式vii所示的化合物的氢谱;图18为实施例15-20中式vii所示的化合物的碳谱;图19为实施例18中式vii所示的不对称产物的hplc的谱图;图20为实施例18中式vii所示的消旋产物的hplc的谱图;图21为实施例9-20中所示的不对称反应的反应机理图。具体实施方式下面结合具体实施方式,进一步阐述本实用。实施例1本发明中配体l1对应的制备方法为:(1)在1,2-二氯乙烷溶剂中,将以下反应方程式中式i所示的化合物、式ii所示的化合物、醋酸催化剂按照1:1.2:0.1的摩尔配比混合均匀,在100度下回流反应10小时,冷却至室温以后,将体系旋干后用乙酸乙酯萃取再用水清洗,后合并有机相,使用无水硫酸钠进行干燥,再浓缩过硅胶柱就可以拿到产物,产物可以重结晶后拿到淡黄色固体(60%产率)。得到式iii所示的化合物(即配体l1),具体的合成路线如下所示:配体l1的对应的氢谱和碳谱数据(如图1、图2所示)如下:1hnmr(400mhz,chloroform-d)δ8.22(s,1h),7.60(d,j=7.8hz,1h),7.33(d,j=8.1hz,1h),7.18(t,j=7.5hz,1h),7.13–7.05(m,2h),4.10(t,j=7.5hz,1h),2.97(qd,j=14.9,7.4hz,2h),1.97(s,3h),1.34(d,j=7.7hz,6h).13cnmr(101mhz,chloroform-d)δ163.63,136.41,127.56,122.44,121.92,119.22,118.77,113.28,111.26,86.31,73.84,28.69,27.15,21.80,14.75.实施例2本发明中配体l2对应的制备方法为:(1)在1,2-含氯甲烷溶剂中,将以下反应方程式中式i所示的化合物、式ii所示的化合物、苯甲酸催化剂按照1:2.0:0.05的摩尔配比混合均匀,在50度下回流反应10小时,分离纯化,冷却至室温以后,将体系旋干后用乙酸乙酯萃取再用水清洗,后合并有机相,使用无水硫酸钠进行干燥,再浓缩过硅胶柱就可以拿到产物,产物可以重结晶后拿到淡黄色固体(70%产率)。得到式iii所示的化合物(即配体l2),具体的合成路线如下所示::配体l2对应氢谱和碳谱数据(如图3、图4所示)如下:(rf=0.3,正己烷:乙酸乙酯=1:1(v/v)).1hnmr(400mhz,chloroform-d)δ8.07(s,1h),7.64(d,j=7.9hz,1h),7.36(d,j=8.1hz,1h),7.20(t,j=7.6hz,1h),7.13(t,j=7.5hz,1h),7.04(s,1h),4.49(t,j=7.4hz,1h),4.18(t,j=8.9hz,1h),3.98(t,j=7.9hz,1h),3.24(dd,j=14.6,5.0hz,1h),2.79(dd,j=14.6,8.8hz,1h),1.98(d,j=1.5hz,3h).13cnmr(101mhz,chloroform-d)δ165.19,136.40,127.63,122.39,122.03,119.35,118.82,111.95,111.26,72.46,66.59,31.59,14.06.hrms(esi)m/zcalcdforc13h15n2o+[m+h+]:215.1179,found:215.1182.实施例3本实施例中配体l2对应的制备方法与实施例2中采用的反应物相同,仅有反应条件的不同,具体如下:在1,2-含氯甲烷溶剂中,将以下反应方程式中式i所示的化合物、式ii所示的化合物、苯甲酸催化剂按照1:1.5:0.08的摩尔配比混合均匀,在80度下回流反应15小时,分离纯化,冷却至室温以后,将体系旋干后用乙酸乙酯萃取再用水清洗,后合并有机相,使用无水硫酸钠进行干燥,再浓缩过硅胶柱就可以拿到产物,产物可以重结晶后拿到淡黄色固体(75%产率),得到式iii所示的化合物(即配体l2)。实施例4本实施例中配体l2对应的制备方法与实施例3中采用的反应物和反应条件的相同,区别仅在于本实施例中是在氩气保护下进行的,得到配体l2的产率为淡黄色固体(80%产率)。实施例5在甲苯溶剂中,将实施例3制备的配体l2作为反应物、碘苯、碘化亚铜、n,n'-二甲基-1,2-环己二胺、磷酸钾按照1:1.1:0.05:0.10:1.2的摩尔配比,在40度下反应24小时,冷却至室温后用乙酸乙酯稀释,过滤后浓缩,经由柱层析方法得到淡黄色油状液体,即得到l3所示的配体(75%产率),具体的合成路线如下所示:配体l3对应氢谱和碳谱数据(如图5、图6所示)如下:(rf=0.2,正己烷:乙酸乙酯=1:1(v/v)).1hnmr(400mhz,chloroform-d)δ7.71–7.65(m,1h),7.56(dd,j=7.9,1.1hz,1h),7.54–7.45(m,4h),7.38–7.29(m,1h),7.26–7.13(m,3h),4.58–4.46(m,1h),4.23(t,j=8.9hz,1h),4.02(t,j=8.5hz,1h),3.28(dd,j=14.6,5.1,1.1hz,1h),2.84(dd,j=14.6,8.8hz,1h),1.99(s,3h).13cnmr(101mhz,chloroform-d)δ165.13,139.82,136.19,129.70,129.16,126.35,126.02,124.25,122.71,120.18,119.30,113.47,110.68,72.50,66.62,31.52,14.14.hrms(esi)m/zcalcdforc19h19n2o+[m+h+]:291.1492,found:291.1493.若保持相同的反应条件下,区别为在氩气保护下进行反应,则配体l3的产率可得到80%。实施例6在二甲苯溶剂中,将实施例4制备的配体l2作为反应物、3,5-双三氟甲基碘苯、碘化亚铜、n,n'-二甲基-1,2-环己二胺、磷酸钾按照1:2.0:0.2:0.4:2.0的摩尔配比,在90度下反应6小时,冷却至室温后用乙酸乙酯稀释,过滤后浓缩,经由柱层析方法得到白色固体,即得到l4所示的配体(77%产率),具体的合成路线如下所示:配体l4对应氢谱和碳谱数据(如图7、图8所示)如下:(rf=0.4,正己烷:乙酸乙酯=1:1(v/v)).1hnmr(400mhz,chloroform-d)δ7.99(d,j=1.5hz,2h),7.84(s,1h),7.73(dt,j=7.7,1.0hz,1h),7.58–7.52(m,1h),7.34(td,j=8.3,7.1hz,1h),7.30–7.24(m,2h),4.62–4.50(m,1h),4.31(t,j=9.4hz,1h),4.04(t,j=8.5hz,1h),3.23(dd,j=14.7,5.8hz,1h),2.92(dd,j=14.7,7.8hz,1h),2.02(d,j=1.3hz,3h).13cnmr(101mhz,chloroform-d)δ165.36,141.31,135.75,133.92,133.58,133.24,132.91,129.84,127.12,125.16,124.41,123.85,123.74,123.70,123.66,123.63,121.69,121.39,119.91,119.49,119.45,119.41,119.38,118.98,115.98,110.00,77.48,77.16,76.84,72.40,66.39,31.39,14.12.19fnmr(376mhz,chloroform-d)δ-62.89(s,ar-cf3).产物的ee值是经由hplc分析,使用的手性柱为:chiralpakiccolumn(正己烷:异丙醇=85:15,1.0ml/min,t=30℃,254nm),tr=4.38min(minor),4.65min(major):>99.5%ee.hrms(esi)m/zcalcdforc21h17n2of6+[m+h+]:427.1240,found:427.1242.若保持相同的反应条件下,区别为在氩气保护下进行反应,则配体l4的产率可得到87%。实施例7在相应的二甲苯溶剂中,将实施例4制备的配体l2作为反应物、五氟碘苯、碘化亚铜、n,n'-二甲基-1,2-环己二胺、磷酸钾按照1:1.5:0.1:0.3:1.5的摩尔配比,在50度下反应14小时,冷却至室温后用乙酸乙酯稀释,过滤后浓缩,经由柱层析方法得到无色油状液体,即得到l5所示的配体(40%产率),具体的合成路线如下所示:配体l5对应氢谱和碳谱数据(如图9、图10所示)如下:hnmr(400mhz,chloroform-d)δ7.72–7.65(m,1h),7.31–7.26(m,1h),7.25–7.20(m,1h),7.11–7.05(m,1h),7.02(s,1h),4.58–4.46(m,1h),4.24(t,j=8.9hz,1h),3.99(dd,j=8.5,7.2hz,1h),3.29–3.18(m,1h),2.86(dd,j=14.8,8.1hz,1h),1.98(d,j=1.3hz,3h).13cnmr(101mhz,chloroform-d)δ165.30,144.80,139.48,136.69,128.78,126.04,123.56,121.15,119.47,115.55,110.32,72.23,66.24,31.19,14.02.19fnmr(376mhz,chloroform-d)δ-144.92,-145.31,-154.42,-154.42,-154.47,-154.53,-160.49,-160.51,-160.55,-160.57,-160.61,-160.63(m,ar-f).hrms(esi)m/zcalcdforc19h14n2of5+[m+h+]:381.1021,found:381.1024.若保持相同的反应条件下,区别为在氩气保护下进行反应,则配体l5的产率可得到56%。实施例8在二甲苯溶剂中,将实施例3制备的配体l2作为反应物、对甲氧基碘苯、碘化亚铜、n,n'-二甲基-1,2-环己二胺、磷酸钾按照1:1.6:0.15:0.3:1.7的摩尔配比,在70度下反应20小时,冷却至室温后用乙酸乙酯稀释,过滤后浓缩,经由柱层析方法得到白色固体,即得到l6所示的配体(82%产率),具体的合成路线如下所示:配体l6对应氢谱和碳谱数据(如图11、图12所示)如下:1hnmr(400mhz,chloroform-d)δ7.67(d,j=7.7hz,1h),7.44(d,j=8.1hz,1h),7.41–7.36(m,2h),7.21(t,j=7.5hz,1h),7.19–7.11(m,2h),7.02(d,j=8.5hz,2h),4.59–4.46(m,1h),4.23(t,j=8.9hz,1h),4.02(t,j=7.9hz,1h),3.88(d,j=0.9hz,3h),3.28(dd,j=14.6,5.0hz,1h),2.83(dd,j=14.6,8.8hz,1h),2.00(d,j=1.5hz,3h).13cnmr(101mhz,chloroform-d)δ165.10,158.18,136.64,132.82,128.74,126.38,125.87,122.48,119.87,119.18,114.81,112.77,110.52,72.52,66.66,55.70,31.52,14.15.hrms(esi)m/zcalcdforc20h21n2o2+[m+h+]:321.1598,found:321.1601.若保持相同的反应条件下,区别为在氩气保护下进行反应,则配体l6的产率可得到88%。实施例9-14本发明还提供了单齿配位噁唑啉配体在不对称反应中的应用,具体包括以下步骤:将式vi所示的底物、亲核试剂对应的n-甲基吲哚、醋酸钯催化剂、(l1-l6)所示的配体中的任一种,o-pba和甲醇溶剂按照1:1.5:0.1:0.3:1.0的摩尔比混合,在室温下搅拌30分钟,随后在60℃的加热块上反应48小时。将反应液冷却至室温,随后将反应体系旋干浓缩后用二氯甲烷萃取三次,再分别用饱和碳酸氢钠,饱和食盐水清洗三次,合并有机相,使用无水硫酸钠干燥,旋干后使用硅胶柱层析分离得到产物,得到式vii所示的化合物,且式vii所示的化合物的ee值为60-86%。其中,实施例9-14中式vii化合物对应的氢谱和碳谱数据(如图13、图14所示)如下:1hnmr(400mhz,chloroform-d)δ9.67(s,1h),8.77(dd,j=7.5,1.6hz,1h),8.75(dd,j=4.3,1.7hz,1h),8.15(dd,j=8.3,1.7hz,1h),7.68(dt,j=7.9,1.0hz,1h),7.59–7.46(m,2h),7.43(dd,j=8.3,4.2hz,1h),7.33–7.24(m,1h),7.20(ddd,j=8.2,6.8,1.2hz,1h),7.07(ddd,j=8.0,6.9,1.1hz,1h),6.90(s,1h),3.73(s,3h),3.19(q,j=7.1hz,1h),2.54(td,j=7.2,2.0hz,2h),2.34–2.17(m,2h),1.44(d,j=7.0hz,3h).13cnmr(101mhz,chloroform-d)δ172.20,148.15,138.45,137.36,136.45,134.73,128.06,127.58,127.28,125.52,121.65,121.58,121.38,119.72,119.66,118.70,116.49,109.34,77.48,77.16,76.84,36.55,33.33,32.75,30.82,22.22.hrms(esi)m/zcalcdforc13h24n3o+[m+h+]:358.1914,found:358.1917.式vii所示化合物的ee值是经由hplc分析,使用的手性柱为:chiralcelad-hcolumn(正己烷:异丙醇=94:6,1.0ml/min,t=30℃,254nm),tr=21.77min(major),24.80min(minor):93:7er.其中,实施例12中式vii所示的不对称产物的hplc的谱图(图15)和式vii所示的消旋产物的hplc的谱图(图16);可以看出配体l4催化不对称反应产生的产率和ee值都很好。不对称反应原理如图21所示,其反应机理为:1.醋酸钯首先选择性与噁唑啉配体结合生成活性钯催化剂(in1),随后与底物进行结合.2.烯烃与pd配位,在o-pba的作用下选择性的解离醋酸根,从而选择性得到关键的手性控制中间体(in2),随后亲核试剂吲哚选择性进攻烯烃,形成中间体(in3),最后通过质子去钯化反应得到最终产物,同时完成催化剂的再生循环.配体的关键作用模式为,配体中的恶唑环上的氮原子与金属pd配位,恶唑环上的甲基起到了区分底物位阻的效果,配体中的吲哚环作用有两方面,第一方面为与pd通过次级轨道作用稳定手性控制中间体,第二方面为提供手性环境,并且会与底物发生位阻作用。吲哚环n-取代基团主要作用就是调节吲哚环的电性,并且会与底物中的喹啉环存在电子堆积作用。配体作用方式的不同:l1配体在主要是因为两个甲基的作用,使配体的结构发生变化,配位角度压缩严重,导致其余底物结合的作用力变小,最终导致其产率的降低和选择性的降低。l2配体主要是由于配位中吲哚的n-h与底物不存在作用力,使形成的中间体in2的构象无稳定,其手性控制能力降低,但是其与pd配位的能力较l1要强,故其收率可以保持较高水平,选择性中等。l3相对于l2,吲哚的n被苯基取代,但是苯基与喹啉环都属于富电子芳环,之间存在排斥作用,故使其稳定性降低,选择性会稍微降低一些。l4相对于l3,将取代基团换成3,5-双三氟甲基苯基,属于缺电子芳环,可以与底物中的喹啉环形成较强的作用力,从而稳定关键中间体的构象,达到最好的手性控制。l5相对于l4,五氟苯基的基团更加缺电子,造成配体与喹啉环的作用太过强,使其结构发生扭曲,造成其手性控制能力大幅降低。l6中对甲氧基苯基是供电子基团,属于富电子芳环,对选择性不利,但是其中的-ome具有一定的配位效果,可以起到一定的稳定构象的作用,从而提高选择性。实施例15-20本发明还提供了单齿配位噁唑啉配体在不对称反应中的应用,具体包括以下步骤:将式vi所示的底物、亲核试剂对应的吲哚、二苯甲酸钯催化剂、(l1-l6)所示的配体中的任一种,o-pba和甲醇溶剂按照1:1.5:0.1:0.3:1.0的摩尔比混合,在室温下搅拌30分钟,随后在60℃的加热块上反应48小时。将反应液冷却至室温,随后将反应体系旋干浓缩后用二氯甲烷萃取三次,再分别用饱和碳酸氢钠,饱和食盐水清洗三次,合并有机相,使用无水硫酸钠干燥,旋干后使用硅胶柱层析分离得到产物,得到式vii所示的化合物,且式vii所示的化合物的ee值为65-90%。实施例对应配体式vii化合物的产率/%式vii化合物的ee值/%实施例15l15465实施例16l29484实施例17l38678实施例18l49590实施例19l59179实施例20l69082对于甲基与乙基的区别:区别在于乙基的基团相对于甲基基团来说,位阻较大,其余配体结合形成的手性空间更加紧凑,而不会发生手性泄露,故其手性选择性相对于甲基而言会提高。通过其他底物的研究,发现取代基团越大,该反应的手性控制越好。其中,实施例15-20中式vii化合物对应的氢谱和碳谱数据(如图17、图18所示)如下:1hnmr(400mhz,chloroform-d)δ9.64(s,1h),8.76(d,j=7.5hz,1h),8.73(dd,j=4.1,1.8hz,1h),8.18–8.10(m,1h),8.08(s,1h),7.67(d,j=7.9hz,1h),7.57–7.45(m,2h),7.42(ddd,j=8.3,4.3,1.4hz,1h),7.36(d,j=8.1hz,1h),7.17(t,j=7.6hz,1h),7.06(t,j=7.5hz,1h),7.02(s,1h),2.91(ddd,j=11.4,9.3,6.0hz,1h),2.59–2.40(m,2h),2.39–2.28(m,1h),2.28–2.14(m,1h),1.83(t,j=7.3hz,2h),0.86(td,j=7.4,1.4hz,3h).13cnmr(101mhz,chloroform-d)δ172.35,148.11,138.35,136.77,136.39,134.62,127.98,127.49,127.17,121.83,121.68,121.62,121.41,119.60,119.08,118.83,116.44,111.34,77.48,77.16,76.84,38.46,36.49,31.20,29.21,12.38.hrms(esi)m/zcalcdforc23h24n3o+[m+h+]:358.1914,found:358.1917.式vii所示化合物的ee值是经由hplc分析,使用的手性柱为:phenomenexcellulose-1column(正己烷:异丙醇=80:20,1.0ml/min,t=30℃,254nm),tr=16.09min(major),18.19min(minor):94:6er.其中,实施例18中式vii所示的不对称产物的hplc的谱图(图19)和式vii所示的消旋产物的hplc的谱图(图20);可以看出配体l4催化不对称反应产生的产率和ee值都很好。实施例21-26本发明提供了单齿配位噁唑啉配体在不对称反应中的应用反应条件与实施例15-20基本相同,区别仅在于本实施例中的反应是在氩气保护下进行的。实施例对应配体式vii化合物的产率/%式vii化合物的ee值/%实施例21l13467实施例22l26486实施例23l35680实施例24l45292实施例25l56081实施例26l66384可以看出,本发明的不对称反应在氮气保护会使化合物的产率略微下降,但会使式vii化合物的ee值的进一步提高,原因在于空气中的少量氧气可能在反应体系中与pd金属配位,从而与手性配体形成竞争,从而使产物的ee值降低。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1