新型靶向苯并咪唑类衍生物抗肿瘤铂(II)和钌(II)配合物及其制备方法与应用与流程
本发明涉及新型靶向苯并咪唑类衍生物金属配合物抗肿瘤药物及其制备领域,尤其是一种新型靶向苯并咪唑类衍生物抗肿瘤铂(ii)和钌(ii)配合物及其制备方法与应用。
背景技术
随着社会的发展、人类寿命的延长,恶性肿瘤对人类生命健康的威胁日益突出。目前,治疗肿瘤的方法有很多,主要包括手术切除、放疗、化疗以及现代生物技术等。其中主要以较为安全、对人体伤害相对较小、对大部分病患适用的化疗为主,而在临床使用的化疗药物中,顺铂、卡铂、奈达铂、奥沙利铂、庚铂、洛铂和米铂等铂类抗肿瘤药物是应用最为广泛的抗肿瘤药物。然而,目前顺铂类药物在卵巢癌、睾丸癌、膀胱癌、大肠癌、肺癌和头颈癌等癌症临床治疗中,仍存在很多缺点,如水溶性差、神经毒性、肾毒性、恶心、呕吐、脱发和乏力等毒副作用和产生的耐药性等。
因此,研发新型高效、低毒、靶向性的铂类和非铂类抗肿瘤化疗药物迫在眉睫。
此外,近年来有许多苯并咪唑衍生物铱(iii)和铑(iii)金属配合物的抗炎、抗溃疡、抗肿瘤、抗高血压、抗氧化、抗细菌、抗增殖、抗病毒和抗真菌特性等在制药领域的应用,但关于苯并咪唑衍生物铂(ii)和钌(ii)配合物的报道却相对比较少。
技术实现要素:
本发明的目的之一是提供一种新型靶向苯并咪唑类衍生物抗肿瘤铂(ii)和钌(ii)配合物。
为了实现上述目的之一,本发明提供的第一技术方案是这样的:一种新型靶向苯并咪唑类衍生物抗肿瘤铂(ii)配合物,其化学结构式如式1所示:
一种新型靶向苯并咪唑类衍生物抗肿瘤钌(ii)配合物,其化学结构式如式2所示:
本发明的目的之二是提供上述两种配合物的制备方法。
为了实现上述目的之二,本发明提供的第二技术方案是这样的:一种如上所述的新型靶向苯并咪唑类衍生物抗肿瘤铂(ii)配合物的制备方法,包括以下步骤:
步骤1:将配体bfcy和二氯·二(二甲基亚砜)合铂(ii)按照物质的量之比为1~5:1~5称取混合,溶于极性溶剂中,得混合溶液;
步骤2:将所述混合溶液于25~80℃条件下反应6~48h,得反应液;
步骤3:将所述反应液过滤,沉淀物经洗涤和干燥后,即得红棕色的目标产物。
一种如上所述的新型靶向苯并咪唑类衍生物抗肿瘤钌(ii)配合物的制备方法,包括以下步骤:
步骤s1:将配体bmcy和二氯·四(二甲基亚砜)合钌(ii)按照物质的量之比为1~5:1~5称取混合,溶于极性溶剂中,得混合溶液;
步骤s2:将所述混合溶液于25~80℃条件下反应6~48h,得反应液;
步骤s3:将所述反应液过滤,沉淀物经洗涤和干燥后,即得红棕色的目标产物。
优选地,所述步骤1和步骤s1中极性溶剂为体积浓度均为1%~99%的甲醇、乙腈、乙醇、二甲基亚砜和/或丙酮,用量为:每1mmol的配体bfcy使用15~80ml极性溶剂;每1mmol的配体bmcy使用10~50ml极性溶剂。
优选地,所述步骤3和步骤s3中洗涤所用溶液均为:乙醚。
优选地,所述步骤3和步骤s3中干燥条件均为:45℃条件下的真空干燥。
两种配合物的合成路线如下所示:
本发明的目的之三是提供上述配合物的应用。
为了实现上述目的之三,本发明提供的第三技术方案是这样的:一种如上所述的新型靶向苯并咪唑类衍生物抗肿瘤铂(ii)和钌(ii)配合物应用于制备抗肿瘤药物。
本发明与现有技术相比,具有以下优点:
本发明以3-(1h-苯并咪唑-2-基)-8-氟-色烯-2-亚基胺(bfcy)或3-(1h-苯并咪唑-2-基)-8-甲基-色烯-2-亚基胺(bmcy)为活性配体,分别与二氯·二(二甲基亚砜)合铂(ii)或二氯·四(二甲基亚砜)合钌(ii)配位反应合成了一种具有抗肿瘤活性的配合物1和2,并考察了它们对t-24、a549、sk-ov-3/ddp、hep-g2、sk-ov-3、hela等人类肿瘤细胞株的增殖抑制活性,结果表明配合物1和2表现出靶向于抑制卵巢癌耐药sk-ov-3/ddp细胞的生长,其ic50值分别为2.08±1.04μm和18.06±0.36μm,其体外抗肿瘤活性远远大于相应的配体bfcy、bmcy和经典的金属基抗癌药物顺铂(75.36±1.65μm);此外,配合物1和2对正常hl-7702肝细胞的毒性很小(ic50>97.0μm)。
综上,配合物1和2表现出优越的体外抗肿瘤活性,具有潜在的药用价值,有望用于各种抗肿瘤药物的制备。
附图说明
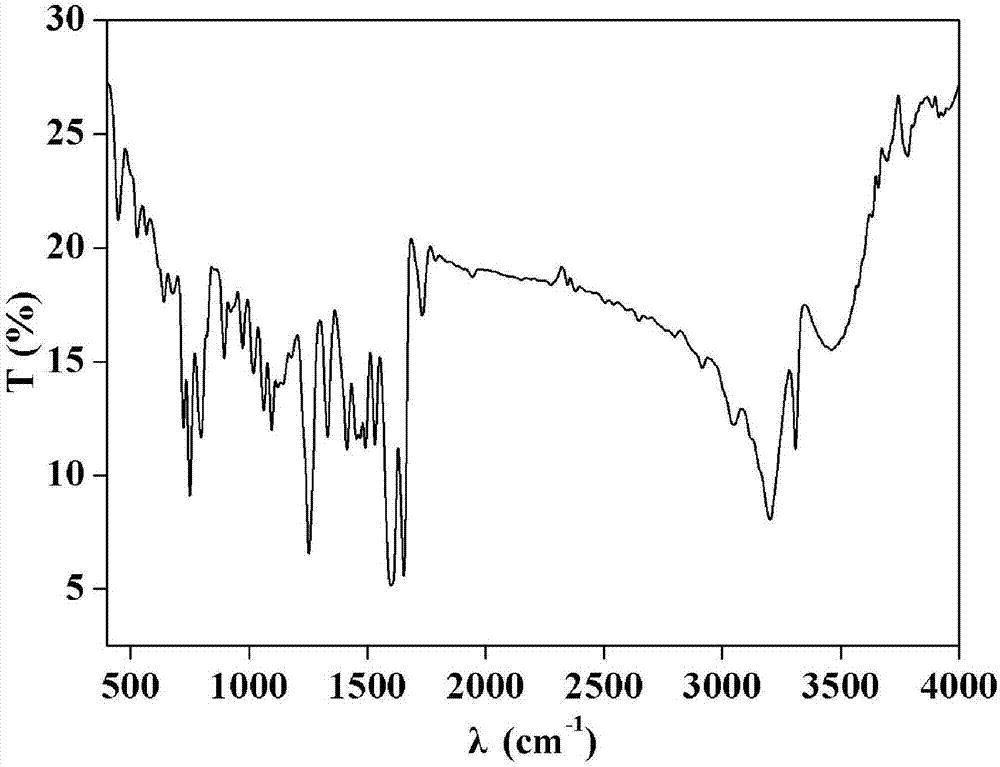
图1为本发明实施例1制得的配合物1的红外谱图;
图2为本发明实施例1制得的配合物1的电喷雾质谱图;
图3为本发明实施例1制得的配合物1的x-射线单晶衍射谱图;
图4为本发明实施例2制得的配合物2的红外谱图;
图5为本发明实施例2制得的配合物2的电喷雾质谱图;
图6为本发明实施例2制得的配合物2的x-射线单晶衍射谱图。
具体实施方式
下面结合具体实施方式,对本发明的权利要求做进一步的详细说明,但不构成对本发明的任何限制,任何在本发明权利要求保护范围内所做的有限次修改,仍在本发明的权利要求保护范围内。
在以下实施例中,涉及的配体bfcy和配体bmcy可参考现有文献(christie,r.m.;etal.dyesandpigments,2000,47:79-89.)进行制备;所涉及的二氯·二(二甲基亚砜)合铂(ii)和二氯·四(二甲基亚砜)合钌(ii)可参考现有文献(al-allaf,t.a.k.;etal.transit.met.chem.,1998,23:403-406.)进行制备。
在以下实施例中,将“二氯·二(二甲基亚砜)合铂(ii)”简写为“cis-ptcl2(dmso)2”;将“二氯·四(二甲基亚砜)合钌(ii)”简写为“cis-rucl2(dmso)4”。
实施例1
向50ml的圆底烧瓶中加入1.0mmol的cis-ptcl2(dmso)2和1.0mmol的配体bfcy,注入30ml的无水甲醇溶液,摇匀后,在45℃下均匀搅拌反应24小时,冷却静置过夜,析出红棕色块状晶体,抽滤,经乙醚洗涤后,在45℃下真空干燥后得红棕色块状晶体,产率90.5%。
对本实施例所得红棕色块状晶体进行以下鉴定:
(1)红外光谱,其谱图如图1所示。
ir(kbr):3461,3310,3202,1654,1600,1533,1493,1455,1414,1332,1097,1063,1019,973,895,798,750,723,528,448cm-1。
(2)电喷雾质谱,其谱图如图2所示。
esi-msm/z:551.4[m-cl+dmso]+,其中m为其分子量。
(3)x-射线单晶衍射谱图,如图3所示。
(4)元素分析结果,如表1所示。
表1元素分析结果
因此,可以确定本实施例所得红棕色块状晶体为铂配合物1。
实施例2
取50ml的圆底烧瓶,分别加入1.0mmol的cis-rucl2(dmso)4和1.0mmol的配体bmcy,注入10ml的丙酮溶液,在65℃下均匀搅拌反应12小时,冷却静置过夜,析出红棕色块状晶体,抽滤,经乙醚洗涤后,在45℃下真空干燥后,得到红棕色块状晶体,产率96.7%。
对本实施例所得红棕色块状进行以下鉴定:
(1)红外光谱,其谱图如图4所示。
ir(kbr):3461,3199,3070,3003,2920,1650,1598,1408,1557,1217,1099,1075,1035,1005,924,763,732,674,424cm-1。
(2)电喷雾质谱,其谱图如图5所示。
esi-msm/z:538.1[m-cl]+,其中m为其分子量。
(3)x-射线单晶衍射谱图,如图6所示。
(4)元素分析结果,如表2所示。
表2元素分析结果
因此,可以确定本实施例所得红棕色块状晶体为钌配合物2。
实施例3
取50ml的圆底烧瓶,分别加入1.0mmolcis-ptcl2(dmso)2和5.0mmol配体bfcy,注入15ml的无水甲醇和5ml二甲基亚砜溶液,摇匀后,在80℃下均匀搅拌反应48小时,冷却静置过夜,析出红棕色块状晶体,抽滤,经乙醚洗涤后,在45℃下真空干燥后,得配合物1,产率85.0%。
实施例4
取50ml的圆底烧瓶,分别加入5.0mmolcis-ptcl2(dmso)2和1.0mmol配体bfcy,注入80ml的无水乙醇溶液,摇匀后,在60℃下均匀搅拌反应24小时,冷却静置过夜,析出红棕色块状晶体,抽滤,经乙醚洗涤后,在45℃下真空干燥后,得配合物1,产率89.0%。
实施例5
取50ml的圆底烧瓶,分别加入1.0mmolcis-rucl2(dmso)4和5.0mmol配体bmcy,注入20ml的乙腈和1ml的水溶液,在25℃下均匀搅拌反应36小时,冷却静置过夜,析出红棕色块状晶体,抽滤,经乙醚洗涤后,在45℃下真空干燥后,得配合物2,产率95.0%。
实施例6
取50ml的圆底烧瓶,分别加入5.0mmolcis-rucl2(dmso)4和1.0mmol配体bmcy,注入50ml的甲醇溶液,在45℃下均匀搅拌反应48小时,冷却静置过夜,析出红棕色块状晶体,抽滤,经乙醚洗涤后,在45℃下真空干燥后,得配合物2,产率85.0%。
为了充分说明配合物1-2在制药中的用途,下面对配合物1-2进行了抗肿瘤活性实验和毒性实验研究。
1.细胞株与细胞培养
本实验选用人膀胱癌细胞t-24、人肺腺癌细胞a549、人卵巢癌sk-ov-3、耐药株sk-ov-3/ddp、人肝癌细胞hep-g2、人宫颈癌细胞hela以及人正常肝细胞hl-77027种人类细胞株。
所有人源细胞株均培养在含100u/ml青霉素、10wt%小牛血和100u/ml链霉素的rpmi-1640培养液内,置37℃含体积浓度5%co2孵箱中培养。
2.待测化合物的配制
所用的配体bfcy、bmcy和配合物1-2的纯度均需≥95%,将它们的dmso储液用生理缓冲液稀释成20μmol/l浓度≤1%的终溶液,测试该浓度下配体bfcy、bmcy和配合物1-2对正常细胞或所选的肿瘤细胞生长的抑制程度。
3.mtt法测定细胞生长抑制实验
(1)取对数生长期的正常细胞或肿瘤细胞,经胰蛋白酶消化后,用含10%小牛血清的培养液配制成浓度为5000个/ml的细胞悬液,以每孔190μl接种于96孔培养板中,使待测细胞密度至1000~10000孔,边缘孔用无菌pbs填充;
(2)5%co2,37℃孵育24h,至细胞单层铺满孔底,每孔加入一定浓度梯度的药物10μl,每个浓度梯度设4个复孔;
(3)5%co2,37℃孵育48小时,倒置显微镜下观察;
(4)每孔加入10μl5mg/ml的pbs溶液,继续培养4h;
(5)终止培养,小心吸去孔内培养液,每孔加入150μl的dmso充分溶解甲瓒沉淀,振荡器混匀后,在酶标仪用波长为570nm,参比波长为450nm测定各孔的光密度值;
(6)同时在调零孔加培养基、mtt和dmso,对照孔加细胞、培养液、mtt、相同浓度的药物溶解介质和dmso。
(7)根据测得的光密度值,即od值,来判断活细胞数量,od值越大,细胞活性越强。利用公式:
计算配体bfcy、bmcy和配合物1-2对所选细胞生长的抑制率,再以bliss法分别计算各受试化合物对所选的各个细胞株的ic50值。
其结果如以下表2所示。
表2对各种细胞株的ic50值(μm)
从ic50活性筛选结果来看,配合物1和2对所选的癌细胞均表现出一定的增殖抑制活性,均远远高于相应的配体bfcy、bmcy和金属盐cis-ptcl2(dmso)2和cis-rucl2(dmso)4的活性,体现出金属钌或铂与配体bfcy、bmcy配位后的协同作用。配合物1和2则对人卵巢癌耐药株sk-ov-3/ddp比较敏感,对其他肿瘤细胞的抑制作用均比较小,它们对sk-ov-3/ddp肿瘤活性的ic50值分别为2.08±1.04μm和18.06±0.36μm,其体外抗肿瘤活性远远大于经典的金属基抗癌药物顺铂的ic50值为75.36±1.65μm。另一方面,配合物1和2对人正常肝细胞hl-7702细胞毒性很小,ic50值均大于97.0μm,表明配合物1和2靶向抑制人卵巢癌耐药株sk-ov-3/ddp的生长,同时还具有较低的肝脏毒性,即配合物配合物1和2具有一定的细胞毒性选择性。
因此,本发明所述的两种新型靶向苯并咪唑类衍生物抗肿瘤铂(ii)、钌(ii)配合物1和2,总体表现出了明显的体外抗肿瘤活性和毒性选择性,具有良好的潜在药用价值,有望用于各种抗肿瘤药物的制备。
- 还没有人留言评论。精彩留言会获得点赞!
- 阻断UBE2M介导Neddylation修饰在肾细胞癌治疗中的应用的制造方法与工艺
- 无载体共组装肿瘤靶向抗癌纳米药物及其制备方法与应用与流程
- 一种用于判断靶向单克隆抗体治疗肿瘤疗效的预测分子的制造方法与工艺
- 一种靶向CD47的免疫检查点抑制剂药物组合物及其制备方法与流程
- 具有淋巴靶向功能的生物自组装纳米晶注射剂及制备方法与流程
- 用于检测肝癌的基因标志物及其用途的制造方法与工艺
- 特异性shRNA筛选及其靶向Ang‑2基因抑制肺癌细胞的验证方法与流程
- 一种多功能自动化生物细胞提取分离控制系统及采用该系统实现的细胞提取分离方法与流程
- 一种融合蛋白及光敏剂复合物及其制备方法和应用与流程
- 具有可视化和显示光斑边界功能的光动力治疗系统的制造方法与工艺
- 一种用于判断靶向单克隆抗体治疗肿瘤疗效的预测分子的制造方法与工艺
- 一种检测肺癌相关基因热点突变的试剂盒及其应用的制造方法与工艺
- 一种为肺癌提供离体个体化药物测试的方法及培养基与流程
- 一种谷胱甘肽/超声双重刺激响应纳米聚合物囊泡、其制备方法及应用与流程
- 一种多重刺激响应交联型聚合物纳米水凝胶、其制备方法及应用与流程
- 一种靶向CD47的免疫检查点抑制剂药物组合物及其制备方法与流程
- 具有淋巴靶向功能的生物自组装纳米晶注射剂及制备方法与流程
- 用于检测肝癌的基因标志物及其用途的制造方法与工艺
- 特异性shRNA筛选及其靶向Ang‑2基因抑制肺癌细胞的验证方法与流程
- 一种具有靶向功能仿病毒结构高分子囊泡及其制备与应用的制造方法与工艺
- 肿瘤靶向核磁共振/荧光双模态成像造影剂及其制备和应用的制造方法与工艺
- pH氧化还原双敏感的PAMAM靶向纳米给药载体及其制备方法与制造工艺
- 具有卵巢肿瘤靶向D构型多肽及其基因递释系统的制造方法与工艺
- 靶向survivin纳米抗体及其制备方法和应用与制造工艺
- 用于PSMA靶向的成像和放射疗法的金属/放射性金属标记的PSMA抑制物的制造方法与工艺
- 一种线粒体靶向的粘度荧光探针及其制备方法和应用与制造工艺
- 一种兼具荧光可视、磁靶向和pH敏多功能的复合空心微球药物载体及其制备方法与制造工艺
- 一种用于修饰微泡的多肽以及靶向GBM的药物制剂的制造方法与工艺
- 白果内酯作为脑靶向增效剂在制备防治脑肿瘤药物的应用的制造方法与工艺
- 机器人辅助精确靶向穿刺软组织目标柔性针夹紧固定装置的制造方法