一种制备2,6-二氯苯甲腈的方法与流程
本发明涉及2,6-二氯苯甲腈制备领域,更具体地说,涉及一种以2,6-二氯氯苄为原料制备2,6-二氯苯甲腈的方法。
背景技术
2,6-二氯苯甲腈又名敌草腈,是一种重要的化学中间体,被广泛应用于除草剂、杀虫剂、医药、染料、高分子材料等领域。
合成2,6-二氯苯甲腈的方法主要分为有机合成法和气相氨氧化法。气相氨氧化法是指在催化剂的作用下,反应物与氨气、空气中的氧气进行氨氧化反应生成2,6-二氯苯甲腈。气相氨氧化法与有机合成法相比,流程简单,能耗低,污染小,产品收率及纯度高,是工业化比较理想的方法。
到目前为止,国内外没有用2,6-二氯氯苄为原料制备2,6-二氯苯甲睛的方法。现有应用气相氨氧化法制备2,6-二氯苯甲睛的方法都是以2,6-二氯甲苯为原料。2,6-二氯甲苯价格高,用其作为反应物来生产2,6-二氯苯甲腈成本昂贵,而2,6-二氯氯苄价格不到2,6-二氯甲苯的一半,用其作为反应物来生产2,6-二氯苯甲腈成本低廉。此外,以2,6-二氯甲苯为原料氨氧化制2,6-二氯苯甲腈的装置多采用流化床,如中国专利zl97109006.8,这是由于氨氧化反应本身放热量大,反应的热不稳定性高,采用固定床无法解决散热及传热问题,只好使用结构复杂的流化床装置,而流化床反应器中流化态质量差,造成产物收率偏低,且催化剂长期消耗,增加生产成本。
因此,如果发明出一种以2,6-二氯氯苄为原料制备2,6-二氯苯甲腈的方法可以很大程度上降低工业生产的成本。如果该方法还能采用固定床反应装置,则更进一步简化工艺条件,提高反应收率,降低生产成本。
技术实现要素:
为了降低工业生产成本,本发明公开了一种以2,6-二氯氯苄(cas号:2014-83-7)为原料制备2,6-二氯苯甲腈的方法。
2,6-二氯氯苄氨氧化制2,6-二氯苯甲腈的反应方程式为:
c7h5cl3+2nh3+o2=c7h3cl2n+nh4cl+2h2o
主要副反应为过度氧化反应,反应方程式为:
2c7h5cl3+6nh3+15o2=6nh4cl+14co2+2h2o
本发明公开了一种制备2,6-二氯苯甲腈的方法,其步骤为:
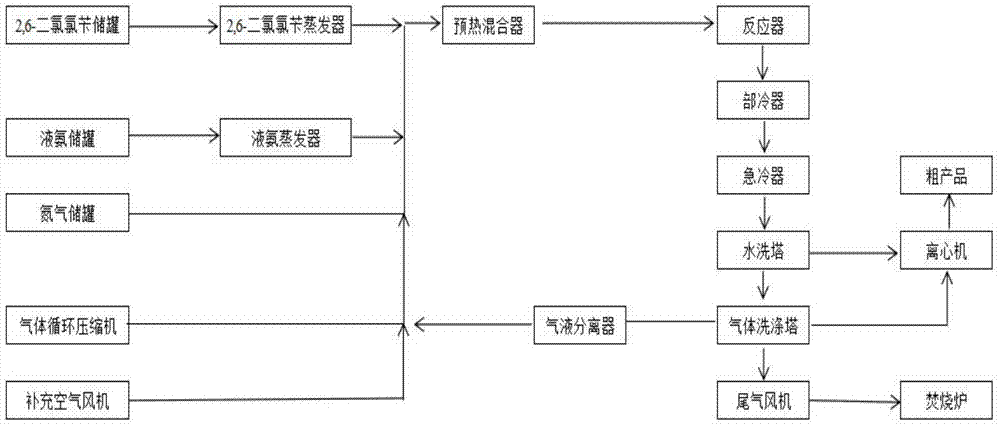
液氨汽化后,与氮气、空气进行气体混合;同时2,6-二氯氯苄通过蒸汽汽化,与上述混合气体一同预热,再于反应器中进行反应,经过降温、水洗、离心脱水得到2,6-二氯苯甲腈粗品。
其中,液氨气化可以经液氨储罐的管内压力自动进入液氨蒸发器等方法来实现;气体混合可以通过气体循环压缩机等来实现;2,6-二氯氯苄可经计量泵进入2,6-二氯氯苄蒸发器,通过中压蒸汽使2,6-二氯氯苄汽化;反应后的产物均为气体,经部冷器(气体冷却器)、急冷器(急冷水喷淋器)降温后,变为固体悬浮颗粒,再进入水洗塔采用急冷水喷淋方式进行水洗,进一步将气体中剩余的2,6-二氯苯甲腈喷淋下来,经过离心机脱水得到粗品。
副产物氯化铵产生量较大,反应器出口温度需要控制在300℃左右,不然易发生管道堵塞。同时,进入水洗塔采用急冷水喷淋方式进行水洗也可以使氯化铵溶于急冷水中,防止氯化铵堵塞管道。
添加氮气的目的是使整个系统内的氧气含量降低,使空气变为贫氧空气。合适的氧气含量有利于抑制过度氧化反应,提高整个反应的选择性。氧气在贫氧空气中的体积含量为6~16%是比较合适的,9~12%更佳,10%左右最佳。
上述方法中2,6-二氯氯苄、氨气、贫氧空气的摩尔比为1:2~10:5~100,优选为1:3~6:10~50,最优选为1:4~6:10~20。
温度在氨氧化强放热反应中一般较难控制,本发明使用的工业反应器的供热和移热是通过一定比例配制的亚硝酸钠和硝酸钾混合物融化后形成的熔盐调节的,通过调节熔盐的温度(简称盐温)来调节反应器的温度,实践证明,当盐温调整到370~410℃,优选380~400℃,最优选390℃时,能够抑制过度氧化反应的发生,提高反应的选择性及收率。
本发明选用的催化剂可为现有的、应用于氨氧化反应制备不同种类的二氯苯甲腈的技术中的已知的催化剂,也可以为本发明公开的一种催化剂(简称dbn-1型催化剂)。其中,使用现有技术中披露的催化剂即可顺利实现以2,6-二氯氯苄为原料制备2,6-二氯苯甲腈的氨氧化反应,但产物的收率及选择性较低。本发明如若使用dbn-1型催化剂,不仅产物的选择性好,收率也很高。
dbn-1型催化剂的制备方法为:
1)活性前驱体的制备
在醇类溶剂中加入钒化合物、磷化合物、钼化合物,搅拌制成悬浊液,加热升温,在反应温度为80℃~160℃、反应压力为0~-40kpa下进行反应,反应4h~6h,冷却后过滤,滤饼于100℃~160℃下干燥24h,再于280℃~400℃下焙烧4h~8h;其中,醇类溶剂为正己醇、异丁醇、苯甲醇中的一种或多种。
2)半成品制备
将步骤1)所得的活性前驱体与载体、助剂、造孔剂、润滑剂混合均匀得到混合物料,其中所述的载体为氧化铝、氧化硅、氧化钛中的一种或多种;所述的助剂为钴化合物和/或铬化合物;也可将混合物料成形并干燥。
3)活化
将步骤2)所得的半成品在氧气含量范围为3%~15%的贫氧空气气氛下进行活化,活化温度为400℃~600℃,活化时间为2h~10h。
本发明的反应器可选用固定床,也可以选用流化床,但在满足本发明的其他参数条件下,使用固定床就能够控制温度和移热,无需再使用结构复杂的流化床,同时,使用固定床还能够避免流化床反应器中流化态质量差,产物收率偏低,催化剂长期消耗,生产成本高的缺陷。
进一步地,为了降低生产成本,防止整个系统中co2含量过高,本发明水洗后的气体经气体洗涤冷却后,75%进入气液分离器回收以降低生产成本,25%使用尾气风机抽进焚烧炉燃烧后放空。回收后的气体与补充空气一起通过循环压缩机循环使用。实践证明,在回收及放空的气体比例为1:3时,反应了的氧气、放空的气体以及补充的空气可以达到平衡。当然,如果换了其他生产设备以实现本发明,回收及放空的气体比例可能会发生一定的变化。
采用本发明可以使低成本的2,6-二氯氯苄通过气相氨氧化法转化为2,6-二氯苯甲腈,降低生产成本;如若采用自制的dbn-1型催化剂,2,6-二氯苯甲腈的收率高,选择性好;还可采用固定床,工艺简单,避免流化床反应器中流化态质量差,产物收率偏低,催化剂长期消耗,生产成本高的问题;如若进一步回收气体则可循环使用,进一步降低成本,非常适合大规模工业生产的需要。
附图说明
图1为实现本方法可以使用的设备及流程示意图。
具体实施方式
下面通过实施例对本发明作进一步阐明。
dbn-1型催化剂的制备:
实施例1:
在带有搅拌、加热和冷凝回流装置的反应釜中,搅拌下加入5000g正己醇和1000g苯甲醇,然后依次加入五氧化二钒630g、磷酸196g、七钼酸铵106g、碳酸钙60g,控制温度在90℃~100℃、-30kpa~-40kpa下进行反应,蒸发的有机溶剂经过水冷凝器冷凝后回流至反应釜中,反应6h后停止反应。将得到的浆料进行固液分离得到滤饼,滤饼在120℃~140℃下干燥18h,然后在300℃~320℃焙烧8h得到活性前驱体。
将活性前驱体加入捏合机中,然后加入氧化铝1524g、硝酸钴87g、田菁粉100g、羧甲基纤维素50g,混合均匀,然后在不断捏合下加入适量的水,直到物料适宜挤条为止。捏合好的物料转入挤条机中挤压成5mm的条,然后用切粒机切成直径5mm的球,球状粒子在110℃~120℃干燥24h,在480℃~500℃下于氧含量9%~11%的气氛下焙烧8h。
实施例2:
在带有搅拌、加热和冷凝回流装置的反应釜中,搅拌下加入5500g正己醇和1100g苯甲醇,然后依次加入偏钒酸铵778g、磷酸138g、四钼酸铵115g、氧化钙50g,控制温度在120℃~130℃、-10kpa~-20kpa下进行反应,蒸发的有机溶剂经过水冷凝器冷凝后回流至反应釜中,反应4h后停止反应。将得到的浆料进行固液分离得到滤饼,滤饼在120℃~140℃下干燥18h,然后在320℃~340℃焙烧6h得到活性前驱体。
将活性前驱体加入捏合机中,然后加入氧化铝1524g、硝酸铬527g、硝酸钴58g、田菁粉100g、羧甲基纤维素50g,混合均匀,然后在不断捏合下加入适量的水,直到物料适宜挤条为止。捏合好的物料转入挤条机中挤压成6mm的条,然后用切粒机切成直径6mm的球,球状粒子在110℃~120℃干燥24h,在500℃~520℃下于氧含量8%~9%的气氛下焙烧8h。
方法实施例:
下述实施例使用的反应器均以固定床反应器为例,等长的反应管,其长度为4000mm,反应器管径为25mm,催化剂采用dbn-1型催化剂,装填高度为3500mm。
下述实施例均使用年产1000吨工业装置,利用氮气先使整个系统内的氧体积含量降低至10%,通过气体循环压缩机使整个系统的空气循环,液氨进入蒸发器汽化后进入循环气体管线与氮气、空气进行气体混合,同时2,6-二氯氯苄进入蒸发器中使2,6-二氯氯苄汽化,再与混合气体进行混合,混合后进入预热混合器一同预热,预热后进入反应器,反应后的气体通过气冷降温至300℃,再经循环冷水喷淋使急冷器内产物降低至80~90℃,产物以固体悬浮颗粒随着急冷水进入水洗塔,水洗塔内部通过急冷水喷淋的方式再把气相中剩余的少量产物喷淋下来,水洗塔底部的固体以间歇采集的方式去离心装置脱水包装。水洗塔中气相去另外一级气体洗涤塔,并把温度降低至40℃以内,75%的气体进入气液分离器回收以降低生产成本,25%的气体使用尾气风机抽进焚烧炉燃烧后放空。回收后的气体与补充空气一起通过循环压缩机循环使用。
实施例3:
三种原料汽体的摩尔比为:2,6-二氯氯苄:氨气:空气=1:3:10,将固定床内盐温调整为390℃,压力为40kpa,加入催化剂,反应12小时。产品的转化率99.5%,选择性为67.2%,收率为66.9%。
实施例4:
三种原料汽体的摩尔比为:2,6-二氯氯苄:氨气:空气=1:3:15,将固定床内盐温调整为390℃,压力为40kpa,加入催化剂,反应12小时。产品的转化率99.5%,选择性为77.0%,收率为76.6%。
实施例5:
三种原料汽体的摩尔比为:2,6-二氯氯苄:氨气:空气=1:4:20,将固定床内盐温调整为390℃,压力为40kpa,加入催化剂,反应12小时。产品的转化率99.5%,选择性为79.7%,收率为79.3%。
实施例6:
三种原料汽体的摩尔比为:2,6-二氯氯苄:氨气:空气=1:4:15,将固定床内盐温调整为390℃,压力为40kpa,加入催化剂,反应12小时。产品的转化率99.5%,选择性为79.5%,收率为79.1%。
实施例7:
三种原料汽体的摩尔比为:2,6-二氯氯苄:氨气:空气=1:5:20,将固定床内盐温调整为390℃,压力为40kpa,加入催化剂,反应12小时。产品的转化率99.5%,选择性为83.9%,收率为83.5%。
实施例8:
三种原料汽体的摩尔比为:2,6-二氯氯苄:氨气:空气=1:6:20,将固定床内盐温调整为390℃,压力为40kpa,加入催化剂,反应12小时。产品的转化率99.5%,选择性为75.4%,收率为75.0%。
实施例9:
三种原料汽体的摩尔比为:2,6-二氯氯苄:氨气:空气=1:6:15,将固定床内盐温调整为390℃,压力为40kpa,加入催化剂,反应12小时。产品的转化率99.5%,选择性为79.8%,收率为79.4%。
实施例10:
三种原料汽体的摩尔比为:2,6-二氯氯苄:氨气:空气=1:5:15,将固定床内盐温调整为390℃,压力为40kpa,加入催化剂,反应12小时。产品的转化率99.5%,选择性为83.5%,收率为83%。
实施例11:
三种原料汽体的摩尔比为:2,6-二氯氯苄:氨气:空气=1:5:15,将固定床内盐温调整为380℃,压力为40kpa,加入催化剂,反应12小时。产品的转化率98.5%,选择性为80.8%,收率为79.6%。
实施例12:
三种原料汽体的摩尔比为:2,6-二氯氯苄:氨气:空气=1:5:15,将固定床内盐温调整为388℃,压力为40kpa,加入催化剂,反应12小时。产品的转化率99.5%,选择性为82.7%,收率为82.3%。
实施例13:
三种原料汽体的摩尔比为:2,6-二氯氯苄:氨气:空气=1:5:15,将固定床内盐温调整为395℃,压力为40kpa,加入催化剂,反应12小时。产品的转化率99.8%,选择性为82.6%,收率为82.4%。
实施例14:
三种原料汽体的摩尔比为:2,6-二氯氯苄:氨气:空气=1:5:15,将固定床内盐温调整为400℃,压力为40kpa,加入催化剂,反应12小时。产品的转化率100%,选择性为78.2%,收率为78.2%。
本发明不局限于上述实施例所述的具体技术方案,凡采用等同替换形成的技术方案均为本发明所保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!