一种多功能抗阿尔茨海默病的苯并咪唑衍生物及其制备方法和应用与流程
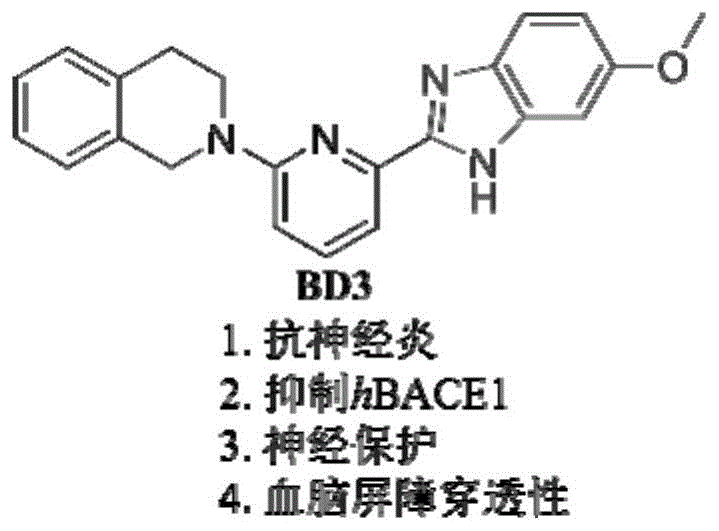
本发明涉及药物设计和药物化学领域,更具体地,涉及一种多功能抗阿尔茨海默症的苯并咪唑衍生物及其制备方法和应用。
背景技术:
阿尔兹海默病(alzheimer’sdisease,ad)俗称老年痴呆,是一种常见的神经系统退行性疾病,症状一般表现为记忆和认知功能的衰退。随着全球人口老龄化加剧及人类寿命延长,ad患者人数正在逐年上升。预计到2030年,全球ad患者人数将达到0.747亿。但目前被fda批准的用于治疗ad的临床药物仅有5种,分别是四种乙酰胆碱酯酶抑制剂:多奈哌齐(donepezil)、利斯的明(rivastigmine)、加兰他敏(galanthamine)和他克林(tacrine),以及一种n-甲基-d-天冬氨酸(nmda)受体拮抗剂美金刚(memantine)。这些药物只能暂时性缓解ad症状且有副作用,而不能减缓或终止疾病进程。因此,寻找新型的抗ad药物和新的疗法具有重要意义。
目前的研究表明,ad的发病机制复杂,涉及到多种病因。其中一种发病假说认为,小胶质细胞主导的神经炎症在ad发病机制中扮演着重要的角色。ad患者大脑中的小胶质细胞处于持续过度激活状态,过度激活状态的小胶质细胞释放大量的炎症因子如一氧化氮(no)等,这些因子会损伤神经元并扩大局部的炎症反应。炎症环境也会促进有毒的淀粉样斑块和神经纤维缠结的形成,神经元受到进一步的损伤,而神经元受损又会反过来激活小胶质细胞,由此形成了小胶质细胞主导的不可控的慢性神经炎。此外,淀粉样级联假说指出,aβ作为元凶诱导了ad的发生。aβ的生成受到一个关键蛋白——bace1的调控。bace1(β-amyloidprecursorproteincleavingenzyme1)又称β分泌酶,是aβ生成过程中的关键限速酶。研究发现,在散发型ad患者大脑中,bace1的活性相对于正常人较高;bace1基因敲除鼠脑内aβ生成减少且未发现有异常。因此,为减少aβ的生成,许多bace1抑制剂被开发用于治疗ad。除了上述提到的两个假说,氧化应激假说认为,ad病理条件下,大量的活性氧和活性氮等物质使细胞内的脂质和蛋白质过氧化,线粒体功能异常,导致神经元的退化和死亡。
随着临床实验药物的失败,有人提出,或许是由于ad病因的复杂多样,才导致这些单一靶点的药物相继折戟。因此,越来越多的人认为,能够同时靶向多个致病因素的“多靶点药物”或许是一个有效治疗ad的策略。
技术实现要素:
本发明针对于现有技术的不足,提供一种多功能抗阿尔茨海默症的苯并咪唑衍生物。
本发明的另一目的在于提供上述苯并咪唑衍生物的制备方法。
本发明的再一目的在于提供上述苯并咪唑衍生物的应用。
本发明采用以下技术方案实现上述技术目的:
一种多功能抗阿尔兹海默病的苯并咪唑衍生物,所述苯并咪唑衍生物的结构如式(i)所示;
其中,r1代表苯并咪唑环六元环上的一个或多个取代基,r2代表四氢异喹啉环六元环上的一个或多个取代基,r1为h、c1~c5的烷基、c1~c5的烷氧基、卤基、羟基或氨基,r2为h、c1~c5的烷基、c1~c5的烷氧基、卤基、羟基或氨基,上述烷基或烷氧基上的氢可被一个或多个c1~c3烷基或卤基取代,x代表c原子或n原子。
苯并咪唑是一种含有两个氮原子的苯并杂环化合物,这一结构存在于多个临床药物中,如抗病毒药物maribavir、降压药telmisartan、治疗胃溃疡药物omeprazole等等。也有一些文献报道苯并咪唑衍生物具有抗炎的活性。此外,四氢异喹啉衍生物被报道具有抗炎、抗氧化、神经保护、改善认知等活性。因此,同时具有苯并咪唑和四氢异喹啉这两个片段的化合物或许会有多种抗ad的活性。再经过化学合成可行性分析,我们选择苯环或者吡啶环将这两个片段连接。
优选地,r1代表苯并咪唑环六元环上的一个取代基,r2代表四氢异喹啉环六元环上的一个或多个取代基。
优选地,r1为h、c1~c3的烷基、c1~c3的烷氧基或卤基。
优选地,r2为h、c1~c3的烷基、c1~c3的烷氧基、羟基、卤素、羟基或氨基。
优选地,r1为h、c1~c3的烷氧基或氟基。
优选地,r2为h、c1~c3的烷氧基、羟基、氨基、甲基、卤素、甲氧基或三氟甲基。
更优选地,所述苯并咪唑衍生物的结构式如下所示:
优选地,所述苯并咪唑衍生物选自下列化合物中的一种:
本发明提供所述的一种多功能抗ad的苯并咪唑衍生物的制备方法,包括如下步骤:通过l-脯氨酸促进的碘化亚铜催化的偶联反应,四氢异喹啉及其衍生物和6-溴吡啶-2-羧酸乙酯反应,得到的这一中间体在碱性条件下水解得到中间体有机羧酸,接着羧酸与邻苯二胺衍生物通过酰胺反应和关环反应,得到苯并咪唑衍生物。
进一步地,制备方法具体包括如下步骤:
s1.
s2.将
当r1不为h时,所述多功能抗阿尔兹海默病的苯并咪唑衍生物的制备方法包括如下步骤:
s1.将
s2.将
s3.将
本发明提供的化合物具有显著的抗神经炎活性、bace1抑制活性、神经保护活性,并且具备较好的透过血脑屏障的能力,可作为治疗老年痴呆的一种先导化合物。
进一步地,本发明提供的化合物通过在bv2细胞模型上抗炎活性筛选,bace1酶活测试以及在ht22细胞上细胞保护测试,发现化合物bd3均表现有活性。并且实验测试发现化合物均可透过血脑屏障。
相对于现有技术,本发明具有如下的优点及效果:
本发明提供的化合物原料易得,制备简单,且是一种多功能抗ad化合物,同时具有抗神经炎活性、bace1酶活抑制活性和细胞保护活性,并可以透过血脑屏障,在制备预防及治疗阿尔茨海默病药物中具备极大的应用前景。
附图说明
图1为化合物bd3的结构示意图。
图2为bd和b系列化合物对谷氨酸诱导ht22细胞死亡的保护作用示意图。
图3为化合物bd3抑制inos蛋白的表达和促炎因子的生成示意图。
图4为bd3通过抑制氧化应激起到细胞保护活性示意图。
具体实施方式
以下结合具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
除非特别说明,本发明所用试剂和材料均为市购。
实施例1:化合物bd1-bd11和b1-b11的合成
按路线一和路线二的合成路线进行化合物的合成。化合物bd1-bd11和b1-b11的反应条件一样,只是取代反应时与中间体四氢异喹啉的反应原料不同,下面以化合物bd1-bd11为例对反应路线进行说明。
通过l-脯氨酸促进的碘化亚铜催化的偶联反应,中间体四氢异喹啉和6-溴吡啶-2-羧酸乙酯反应,得到中间体1,接着中间体1在碱性条件下水解得到中间体2;中间体有机羧酸2与原料邻苯二胺衍生物通过酰胺反应和关环反应,最终得到bd系列终产物bd1-bd9。对于bd系列中的另外两个终产物bd10和bd11,由于中间体——6位取代的四氢异喹啉衍生物无法购买,所以在合成这两个终产物时,需要先经过两步反应合成中间体5和6:原料5-氟-1-茚酮或者5-甲氧基-1-茚酮在酸性条件下,与叠氮化钠经过aube-schmidt重排反应生成酰胺,然后中间体3和4被氢化铝锂还原,分别得到6位取代的四氢异喹啉衍生物5和6。
路线一:bd系列化合物bd1-bd11的合成路线;
其中,条件如下:(a)k2co3,cui,l-proline,dmso,80℃,16h;(b)lioh,thf,reflux,3h;(c)tbtu,dipea,dmf,r.t.,6h,thenacoh,reflux,overnight;(d)meso3h,nan3,dcm,r.t.,overnight;(e)allih4,thf,reflux,4h.
路线二:b1-b11的合成路线:
其中,条件如下:(a)k2co3,cui,l-proline,dmso,80℃,16h;(b)lioh,thf,reflux,3h;(c)tbtu,dipea,dmf,r.t.,6h,thenacoh,reflux,overnight.
具体步骤如下:
(1)中间体1及其他甲酸乙酯衍生物的合成
向装有原料6-溴吡啶-2-羧酸乙酯(4.32g,20mmol)和1,2,3,4-四氢异喹啉(5ml,40mmol)的耐压管中,加入碳酸钾(8.34g,60mmol)、碘化亚铜(0.8g,4mmol)和l-脯氨酸(0.92g,8mmol),再加入适量的二甲基亚砜dmso作为反应溶剂,氮气保护,油浴加热到80℃,磁力搅拌反应16h。反应结束后,向反应体系中加入适量水,然后用乙酸乙酯萃取(3×150ml),而后在减压下旋干,残液经柱层析分离纯化(石油醚:乙酸乙酯=5:1)得到白色油状物中间体1(3.94g,70%)。
中间体7的合成由中间体5(2.45g,15mmol)和6-溴吡啶-2-羧酸乙酯(2.30g,10mmol)按上述方法合成,柱层析分离纯化(石油醚:乙酸乙酯=8:1)得到白色油状物中间体7(2.34g,75%)。
中间体8的合成由中间体6(2.26g,15mmol)和6-溴吡啶-2-羧酸乙酯(2.30g,10mmol)按上述方法合成,柱层析分离纯化(石油醚:乙酸乙酯=8:1)得到黄色油状物中间体8(2.16g,72%)。
中间体11的合成由3-碘苯甲酸乙酯(5.52g,20mmol)和1,2,3,4-四氢异喹啉(5ml,40mmol)按上述方法合成,柱层析分离纯化(石油醚:乙酸乙酯=10:1)得到白色油状物中间体11(4.21g,75%)。
中间体13的合成由3-碘苯甲酸乙酯(2.76g,10mmol)和中间体5(2.45g,15mmol)按上述方法合成,柱层析分离纯化(石油醚:乙酸乙酯=20:3)得到白色油状物中间体13(2.17g,70%)。
(2)中间体2及其他羧酸衍生物的合成
称取中间体1(2.82g,10mmol),加入50ml四氢呋喃,氢氧化锂(1.19g,50mmol)用乙醇:水(5:1)混合物溶解后加入到圆底瓶中,回流反应3h。反应结束后,将反应液减压旋干,加入少量冰水溶解,然后冰浴下用稀盐酸缓慢调节ph至弱酸性,直至有固体析出。将析出的固体抽滤,干燥,得到中间体2为淡黄色固体(2.33g,92%)。
中间体12的合成由中间体11(2.81g,10mmol)按上述方法合成,得到淡黄色固体12(1.63g,65%)。
中间体9的合成由中间体7(1.56g,5mmol)按上述方法合成,得到淡黄色固体9(0.99g,70%)。
中间体10的合成由中间体8(1.50g,5mmol)按上述方法合成,得到黄色固体10(1.08g,80%)。
中间体14的合成由中间体13(1.55g,5mmol)按上述方法合成,得到淡黄色固体14(1.13g,80%)。
(3)中间体3和4的合成
原料5-甲氧基-1-茚酮(4.30,26.6mmol)溶解在40ml二氯甲烷和40ml甲基磺酸的混合液中,冰浴冷却到0℃后,将叠氮化钠(3.46g,53.2mmol)加入反应瓶中,室温搅拌过夜。反应结束后,冰浴下向反应体系中缓慢加入20%氢氧化钠溶液调节ph至弱碱性,然后用二氯甲烷萃取,有机层用无水硫酸镁干燥,减压旋干,柱层析分离纯化(石油醚:乙酸乙酯=1:2)得到白色固体中间体3(2.82g,60%)。中间体4的合成由原料5-氟-1-茚酮(4.00g,26.6mmol)按上述方法合成,得到白色固体4(2.32g,53%)。
(4)中间体5和6的合成
中间体3(1.77g,10mmol)溶于50ml干燥的四氢呋喃中,0℃下加入1mol/l氢化铝锂的四氢呋喃悬液(20ml),回流反应4h。反应结束后,冰浴下用30%氢氧化钠溶液淬灭反应,硅藻土过滤,并用甲醇洗涤,合并滤液在减压下旋干,粗产物经柱层析纯化(乙酸乙酯:甲醇=10:1)得到白色油状物5(0.81g,50%)。中间体6的合成由中间体4(1.65g,10mmol)按上述方法合成,得到白色油状物6(0.95g,63%)。
(5)终产物bd系列和b系列化合物的合成通法
将有机酸(1mmol)溶于5mln,n-二甲基甲酰胺中,冰浴下加入n,n-二异丙基乙胺(0.21ml,1.2mmol),然后加入o-苯并三氮唑-n,n,n',n'-四甲基脲四氟硼酸酯tbtu(385mg,1.2mmol)搅拌30分钟,之后,加入相应的邻苯二胺衍生物(1.1mmol),室温下反应6h。然后冰水淬灭反应,用乙酸乙酯萃取,有机层用无水硫酸镁干燥,减压旋干,然后将旋干得到的固体溶于10ml冰乙酸中,回流反应过夜。反应结束后,冷却至室温,用饱和的碳酸氢钠溶液调节ph至中性,乙酸乙酯萃取,有机层干燥,减压旋干,经柱层析分离纯化得到相应的终产物。
化合物bd1-bd11和b1-b11的结构、外观和核磁共振氢谱数据如表1所示。
表1化合物bd1-bd11和b1-b11的结构、外观和核磁共振氢谱数据
实施例2实施例1所得的22个化合物的抗ad活性评价
对以上合成得到的22个苯并咪唑衍生物进行抗ad活性评价,主要包括以下三种活性测试:对炎症因子no生成的抑制活性、对bace1的体外酶活抑制活性、对谷氨酸诱导的ht22细胞死亡的保护活性等。
1.no抑制活性测试
(1)细胞培养。体外培养bv2细胞(小鼠小胶质细胞)。使用含有10%胎牛血清,链霉素(100微克/毫升),青霉素(100单位/毫升)的dmem培养基,在37℃,5%二氧化碳浓度条件下进行常规维持培养和传代。
(2)药物干预。取处于对数生长期的细胞,消化,离心,重悬后按每孔100μl,每孔细胞数4×104个接种于96孔板中,培养24小时后吸除原培养液,加入不含血清的dmem培养基80μl,30分钟后加入不同浓度的目标化合物10μl,最终的浓度为(0.5、1、5、10、25、50μm),空白对照组和lps单独处理组不加,预处理30分钟后,各组加入lps(10μg/ml)10μl,即最终浓度为1μg/ml,放入培养箱中继续培养24h后检测。
(3)griess法检测目标化合物抑制no实验。按试剂盒说明测定培养液中的no水平,主要步骤如下:
①按50μl/孔,在96孔板中加入标准品及样品。
②按50μl/孔,在各孔中加入室温griess试剂i。
③按50μl/孔,在各孔中加入室温griess试剂ii。
④540nm测定吸光度。
(4)结果处理。按照公式:(fl-fc)/(fl-f0)×100%,其中,fl是lps处理组的od值,fc是化合物处理组的od值,f0是未处理细胞组的od值,计算各化合物在不同浓度下对no的相对抑制率。以相对抑制率为纵坐标,化合物浓度为横坐标绘制各化合物对no生成的抑制曲线图,并计算各化合物的半数抑制剂量(ic50)以评价各化合物对no生成的抑制活性。
化合物抑制no生成的ic50如表2所示。其中除了6个化合物(bd7、bd10、b3、b8、b10、b11)抑制no的ic50值大于10μm外,其他16个化合物的ic50值均处于1~10μm,并且优于白藜芦醇(ic50=11.1μm)。
表2化合物bd1-bd11和b1-b11抑制no生成的ic50、bace1抑制率和透过bbb能力
2.bace1酶活抑制活性测试
(1)用醋酸钠缓冲液把bace1蛋白稀释至20ng/μl,底物稀释至25μm,化合物稀释至200μm(含2%dmso)。
(2)10μl化合物与50μl的bace1蛋白溶液加入到96孔板混匀后,在室温孵育20分钟;标准对照孔中加入10μl含2%dmso的醋酸钠缓冲液。
(3)往96孔板中加入40μl底物,混匀,立即在ex=345nm和em=500nm处检测荧光吸收,作为空白(因为底物和化合物本身都会有荧光吸收,计算抑制率时应扣除背景)。
(4)37℃反应1h后,再次检测荧光吸收。
(5)数据处理。bace1抑制率(%)=(f对照-f样品)/(f对照-f空白)×100%,其中f是测得的荧光值。
所合成的苯并咪唑衍生物在20μm浓度下对bace1的抑制率如表2所示,该实验中以bace1抑制剂mk-8931为阳性对照。从表2可以看到,5个化合物b3、bd3、bd5、bd8和bd9对bace1的酶活抑制率在60%以上,其余化合物几乎无抑制活性。
3.谷氨酸诱导ht22细胞死亡的保护活性测试
(1)细胞培养。体外培养ht22细胞(小鼠海马神经元细胞)。使用含有10%fbs,链霉素(100微克/毫升),青霉素(100单位/毫升)的dmem培养基,在37℃,5%二氧化碳浓度条件下进行常规维持培养和传代。
(2)化合物干预。ht22细胞按5000个/孔接种于96孔板中,培养24h后,除去旧培养基,加入含有不同浓度化合物的新培养基和2mm谷氨酸,继续培养24小时后,用mtt法检测细胞的存活率。
(3)数据处理。细胞存活率(%)=(od样品-od空白)/(od对照-od空白)×100%
所合成的苯并咪唑衍生物在5μm和1μm浓度下对谷氨酸(glu)诱导的ht22细胞死亡的保护作用如图3所示。由图3可以看出,谷氨酸处理24h后,细胞的存活率仅有40%左右,5μmb3、b8、b10和b11分别处理后,细胞存活率比谷氨酸处理后的细胞存活率明显提高,说明这4个化合物在5μm时对ht22细胞有显著的保护作用。bd系列化合物中有6个化合物(bd1、bd2、bd3、bd5、bd6、bd11)在5μm时有显著的细胞保护活性,细胞存活率都在60%以上,但这一系列化合物在1μm时的细胞存活率几乎与谷氨酸处理后的细胞存活率相当,均无细胞保护作用。
实施例3实施例1所得的22个化合物的pampa-bbb实验
1.试剂准备
(1)配制2%的猪脑提取物(porcinebrainlipid,pbl):称取20mg猪脑提取物加入1ml的正十二烷,使其充分溶解。使用前配制。
(2)配制50mm的pbs:称取1.36gk2hpo4于200ml超纯水中,koh调ph至7.4。
(3)配制5mg/ml对照药物储备液:称取5mg对照药物加入1mldmso溶解,-20℃保存。
(4)配制100μg/ml待测样品液:取储备液20μl于1.5ml的ep管中,加入980μl缓冲液(ph7.4pbs:乙醇=70:30)。
2.操作步骤
(1)小心吸取4μl2%的猪脑提取液加入作为给药池的96孔板疏水膜上。
(2)迅速吸取200μl待测样品液于96孔板中作为给药池,接收池加入300ul缓冲液(ph7.4pbs:乙醇:dmso=68:30:2)。
(3)小心的将给药池平放于接收池上,并使膜与接收液充分接触。
(4)室温静止10小时,小心移去给药池。用多功能酶标仪测试接收池内化合物在其最大吸收峰处的od值。
(5)吸取200μl待测样品液于300μl缓冲液(ph7.4pbs:乙醇:dmso=68:30:2)中混匀,作为理论平衡溶液,测其化合物在最大吸收峰处的od值。
(6)以含2%的dmso缓冲液作为空白对照。
(7)根据公式计算出pe值
pe=-vd×va/[(vd+va)a×t]×ln(1-od接收池内待测样品/od理论平衡溶液待测样品)
vd为给药池体积,vd为接收池体积,a为渗透面积,t为渗透时间。当pe>4.7时既表示化合物具有较好的透过血脑屏障能力。如下所示:
实施例1所得的22个化合物的pe值如表2所示,化合物的pe均大于4.7,说明化合物均可透过血脑屏障。
以上完成了对所合成的苯并咪唑衍生物在抑制no生成、抑制谷氨酸诱导ht22细胞死亡和抑制bace1酶活这三方面的活性测试,综合考虑化合物的活性,其中化合物bd3在这三种活性测试中都表现有活性,其抑制no生成的ic50=5.07μm,20μm时对bace1抑制率达到65.7%,5μm时能够完全抑制谷氨酸诱导的细胞毒性,并且可以透过血脑屏障,因此,我们选择化合物bd3进行了初步的抗ad机制研究。
实施例4实施例1所得的化合物bd3抗神经炎的机理研究
当bv2细胞受到脂多糖lps刺激时,细胞内的诱导型一氧化氮合酶(induciblenitricoxidesynthase,inos)以及促炎因子白介素1β(il-1β)和肿瘤坏死因子tnf-α会增加。因此,通过westernblot、elisa、qrt-pcr实验检测bd3对细胞内inos、il-1β和tnf-α表达的抑制效果,结果如图4所示。化合物bd3均表现有抑制活性,并且呈现浓度依赖性。因此,bd3通过抑制inos、il-1β和tnf-α的表达起到了显著的抗神经炎活性。
实施例5实施例1所得的化合物bd3抑制谷氨酸诱导ht22细胞死亡的保护机理研究
gsh是体内一种重要的非酶抗氧化剂和自由基清除剂,可清除o2-、h2o2等。gsh水平的高低决定着机体抗氧化能力的强弱。gsh竭耗是谷氨酸导致细胞死亡的主要因素之一,所以我们利用试剂盒对细胞内gsh含量进行检测。结果如图4中(a)所示。从图中可以看出,谷氨酸处理后,细胞内gsh的含量明显下降;细胞经过化合物bd3处理后,当浓度为1μm时,gsh水平与谷氨酸单独处理组并无太大差异;当bd3浓度提高为3μm时,gsh水平几乎能够恢复到正常细胞的水平,说明在3μm时,bd3可以抑制谷氨酸造成的gsh竭耗。谷氨酸诱导ht22细胞产生氧化应激从而导致细胞死亡,如图4中(b)和(c)所示,谷氨酸处理后,细胞内ros的含量明显升高;细胞经过化合物bd3处理后,当bd3浓度为3μm时,荧光强度明显减弱,说明bd3可以显著抑制谷氨酸诱导的ros生成;而在1μm时,荧光强度与谷氨酸单独处理组并无明显差异,说明bd3在1μm时不能抑制ros生成。这一数据结果与gsh的数据结果互相吻合。综合以上数据,可以得到结论:bd3在3μm时能够抑制谷氨酸诱导的ht22细胞死亡,进一步的实验结果表明,该化合物在3μm时通过抑制ros生成和提高gsh含量,减弱了谷氨酸诱导的氧化应激,从而起到了细胞保护活性。
上述实验结果表明本发明所述化合物为多功能抗ad活性化合物,不仅能够抑制神经炎症和bace1酶活,还具有神经保护活性,可作为治疗ad的先导化合物。
- 还没有人留言评论。精彩留言会获得点赞!