一种液态奶中微生物总DNA的提取方法与流程
本发明涉及一种dna的提取方法,具体涉及一种液态奶中微生物总dna的提取方法。
背景技术:
液态奶样品中微生物多样性分析日显重要,传统的分离培养技术,菌种形态检测技术等,一方面受到模拟培养环境上的挑战,另一方面受到了准确性及灵敏性的限制。随着高通量测序技术的发展及测序成本的大幅度降低,dna水平上检测牛奶中微生物的组成,有着高灵敏度,较高准确度的优势。
16s扩增子测序技术是研究环境中原核微生物多样性及群落组成差异的高通量测序技术之一,通过提取环境或者食品样品的dna,选择合适的通用引物扩增16s的目标区域,通过检测目标区域的序列变异和丰度,来研究微生物多样性分布及差异分布。
技术实现要素:
为了解决现有技术存在的问题,本发明提供了一种液态奶中微生物总dna的提取方法,其具有耗时短,提取的dna质量更纯,更高等特点。
本发明的目的是提供一种液态奶中微生物总dna的提取方法。
根据本发明的具体实施方式的液态奶中微生物总dna的提取方法,所述提取方法包括以下步骤:
(1)样品处理:室温均质牛奶样品,取1.0ml牛奶样品进行离心,弃掉上清液,得到沉淀物;
(2)细胞裂解:在步骤(1)得到的所述沉淀物中enzymaticlysisbuffer和lysozyme混匀后孵育,得到裂解液;
(3)纯化dna:在步骤(2)得到的所述裂解液中加入酚氯仿进行抽提,旋涡振荡,直至白色悬浊液出现,室温孵育5min,然后离心取上清液;
(4)回收dna:在步骤(3)得到的所述上清液中加入glycogen、醋酸钠和异丙醇,混匀,-20℃孵育30min,离心弃掉上清液,并同时保留沉淀;
(5)溶解dna:在步骤(4)得到的所述沉淀中加入75%无水乙醇进行洗涤,弃掉上清液,将沉淀室温晾干3-5min;加入20μltebuffer或者ddh2o,室温孵育8-10min后,轻弹混匀或者轻微涡旋混匀,即可获得检测样品。
其中,tebuffer或者ddh2o为洗脱缓冲液。得到的检测样品也可以保存于-80℃的冰箱以便后续使用。
根据本发明的具体实施方式的液态奶中微生物总dna的提取方法,其中,步骤(1)中,将所述牛奶样品室温13000xg离心5min,沉淀样品,弃掉上清液,得到沉淀物。
根据本发明的具体实施方式的液态奶中微生物总dna的提取方法,其中,步骤(2)中,在所述沉淀物中加入500μl的enzymaticlysisbuffer和1μl的lysozyme混匀后孵育,加入3μl蛋白质k,孵育1h,得到裂解液。
据本发明的具体实施方式的液态奶中微生物总dna的提取方法,进一步的,在所述沉淀物中加入500μl的enzymaticlysisbuffer和1μl的lysozyme混匀后孵育,加入3μl蛋白质k,孵育1h,然后玻璃珠研磨10min,得到裂解液。玻璃珠研磨是玻璃珠的随机碰撞,目的是物理性破坏菌体肽聚糖细胞壁,进一步降解细胞壁。加入玻璃珠后震荡10min,由于剧烈震荡造成试管内有较多气泡,为了避免补液时液体外溢造成交叉污染,可以室温条件下,13000xg离心3min,消除气泡的影响。
根据本发明的具体实施方式的液态奶中微生物总dna的提取方法,进一步的,在所述沉淀物中加入500μl的enzymaticlysisbuffer和1μllysozyme,上下吹打混匀,37℃孵育1h,后,加入3μl蛋白质k到裂解液中,55℃热孵育1h,得到重悬浮液,将所述重悬浮液转移到一个含有0.8-1g玻璃珠子的离心管中振荡10min,得到裂解液。
根据本发明的具体实施方式的液态奶中微生物总dna的提取方法,进一步的,得到第一次裂解液后,将所述第一次裂解液13000xg离心3min,再向所述第一次裂解液中补加300μlsdslysisbuffer。
根据本发明的具体实施方式的液态奶中微生物总dna的提取方法,其中,步骤(3)中,所述上清液中加入1倍体积的酚氯仿进行抽提,旋涡振荡,直至白色悬浊液出现,室温孵育5min,然后在4℃下,16000xg离心10min,取上清液。
根据本发明的具体实施方式的液态奶中微生物总dna的提取方法,其中,步骤(3)中,向步骤(2)得到的所述上清液中加入0.2倍体积的10m醋酸铵,冰上孵育5min,然后在25℃下,13000xg离心10min,取第二次上清液,加入1倍体积酚氯仿进行抽提,旋涡振荡,直至白色悬浊液出现,室温孵育5min,然后离心取上清液。
根据本发明的具体实施方式的液态奶中微生物总dna的提取方法,其中,步骤(4)中,所述上清液中加入1μl的glycogen、1/10体积的3m醋酸钠和1倍体积异丙醇,所述醋酸钠的ph为5.2,上下颠倒混匀5-6次,-20℃孵育30min,4℃,16000xg离心15min,弃掉上清液,并同时保持沉淀在离心管中。
根据本发明的具体实施方式的液态奶中微生物总dna的提取方法,其中,步骤(5)中,在所述沉淀中加入新鲜配制的75%无水乙醇洗涤两次,弃掉上清液,将沉淀室温晾干3-5min;加入20μltebuffer或者ddh2o,室温孵育8min后,轻弹混匀或者轻微涡旋混匀,即可获得检测样品。
根据本发明的具体实施方式的液态奶中微生物总dna的提取方法,进一步的,所述洗涤两次具体为:在所述沉淀中加入新鲜配制的75%无水乙醇,上下颠倒混匀使白色沉淀完全漂浮在洗液中,4℃,16000xg离心5min,弃掉上清液,再加入新鲜配制的75%无水乙醇重复洗涤一次,去掉上清液,保留沉淀。
与现有技术相比,本发明具有如下有益效果:
(1)本发明的液态奶中微生物总dna的提取方法,耗时短:整个流程只需2小时;
(2)步骤少:转移2-3次,只使用2-3次新离心管;
(3)无裂解液浪费,裂解后,直接进行有机试剂抽提,减少dna损失;
(4)高质量提取:提取的dna质量比传统方法更纯、质量更高,提取dna的量比传统方法更大,可用于后续研究;
(5)可重复性、易操作。
附图说明
图1显示不同提取方法得到的基因组dna样品的1%琼脂糖凝胶电泳对比图;
图2显示本发明的实施例3和实施例4得到的基因组dna样品经pcr扩增后,进行琼脂糖凝胶电泳检测的对比图;
图3显示本发明的实施例3得到的基因组dna的agilent2100定性检测结果;
图4显示本发明的实施例4得到的基因组dna的agilent2100定性检测结果。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将对本发明的技术方案进行详细的描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施方式,都属于本发明所保护的范围。
本发明中所用的试剂,包括enzymaticlysisbuffer、lysozyme、tebuffer、glycogen、醋酸钠和异丙醇等均采购自生工生物工程(上海)股份有限公司。
实施例1
本实施例提供了一种液态奶中微生物总dna的提取方法,所述提取方法包括以下步骤:
(1)样品处理:室温均质牛奶样品,取1.0ml牛奶样品进行离心,弃掉上清液,得到沉淀物;
(2)细胞裂解:在步骤(1)得到的所述沉淀物中加入enzymaticlysisbuffer和lysozyme混匀后孵育,得到裂解液。
(3)纯化dna:在步骤(2)得到的所述裂解液中加入酚氯仿进行抽提,旋涡振荡,直至白色悬浊液出现,室温孵育5min,然后离心取上清液;
(4)回收dna:在步骤(3)得到的所述上清液中加入glycogen、醋酸钠和异丙醇,混匀,-20℃孵育30min,离心弃掉上清液,并同时保持沉淀;
(5)溶解dna:在步骤(4)得到的所述沉淀中加入75%无水乙醇进行洗涤,弃掉上清液,将沉淀室温晾干3min;加入20μltebuffer,室温孵育8-10min后,轻弹混匀或者轻微涡旋混匀,获得检测样品保存于-80℃的冰箱以便后续使用。
实施例2
本实施例提供了一种液态奶中微生物总dna的提取方法,所述提取方法包括以下步骤:
(1)样品处理:室温均质牛奶样品,取1.0ml牛奶样品室温13000xg离心5min,沉淀样品,弃掉上清液,得到沉淀物;
(2)细胞裂解:在步骤(1)得到的所述沉淀物中加入500μl的enzymaticlysisbuffer和1μllysozyme,上下吹打混匀,37℃孵育1h,后,加入3μl蛋白质k到裂解液中,55℃热孵育1h,得到重悬浮液,将所述重悬浮液转移到一个含有1g玻璃珠子的离心管中振荡10min,得到裂解液;玻璃珠研磨是玻璃珠的随机碰撞,目的是物理性破坏菌体肽聚糖细胞壁,进一步降解细胞壁;
(3)纯化dna:在步骤(2)得到的所述裂解液中加入0.2倍体积的10m醋酸铵进行盐析,冰上孵育5min,然后在25℃下,13000xg离心10min,取第二次上清液加入1倍体积的酚氯仿进行抽提,旋涡振荡,直至白色悬浊液出现,室温孵育5min,然后离心取上清液;所述上清液中加入1倍体积的酚氯仿进行抽提,旋涡振荡,直至白色悬浊液出现,室温孵育5min,然后在4℃下,16000xg离心10min,取上清液;低浓度盐离子可以促进蛋白溶解,称为盐析;
(4)回收dna:在步骤(3)得到的所述上清液中加入1μl的glycogen、1/10体积的3m醋酸钠和1倍体积异丙醇,所述醋酸钠的ph为5.2,上下颠倒混匀5次,-20℃孵育30min,4℃,16000xg离心15min,弃掉上清液,并同时保持沉淀在离心管中;
(5)溶解dna:在步骤(4)得到的所述沉淀中加入新鲜配制的75%无水乙醇上下颠倒混匀使白色沉淀完全漂浮在洗液中,4℃,16000xg离心5min,弃掉上清液,再加入新鲜配制的75%无水乙醇重复洗涤一次,去掉上清液,保留沉淀,将沉淀室温晾干5min;加入20μl的ddh2o,室温孵育8min后,轻弹混匀或者轻微涡旋混匀,即可获得检测样品。
实施例3
本实施例提供了一种液态奶中微生物总dna的提取方法,所述提取方法包括以下步骤:
(1)样品处理:室温均质牛奶样品,取1.0ml牛奶样品室温13000xg离心5min,沉淀样品,弃掉上清液,得到沉淀物;
(2)细胞裂解:在步骤(1)得到的所述沉淀物中加入500μl的enzymaticlysisbuffer和1μllysozyme,上下吹打混匀,37℃孵育1h,后,加入3μl蛋白质k到裂解液中,55℃热孵育1h,得到重悬浮液,将所述重悬浮液转移到一个含有0.8g玻璃珠子的离心管中振荡10min,得到裂解液;玻璃珠研磨是玻璃珠的随机碰撞,目的是物理性破坏菌体肽聚糖细胞壁,进一步降解细胞壁;
(3)纯化dna:在步骤(2)得到的所述裂解液中加入1倍体积的酚氯仿进行抽提,旋涡振荡,直至白色悬浊液出现,室温孵育5min,然后离心取上清液;所述上清液中加入1倍体积的酚氯仿进行抽提,旋涡振荡,直至白色悬浊液出现,室温孵育5min,然后在4℃下,16000xg离心10min,取上清液;
(4)回收dna:在步骤(3)得到的所述上清液中加入1μl的glycogen、1/10体积的3m醋酸钠和1倍体积异丙醇,所述醋酸钠的ph为5.2,上下颠倒混匀6次,-20℃孵育30min,4℃,16000xg离心15min,弃掉上清液,并同时保持沉淀在离心管中;
(5)溶解dna:在步骤(4)得到的所述沉淀中加入新鲜配制的75%无水乙醇洗涤两次,弃掉上清液,将沉淀室温晾干5min;加入20μltebuffer,室温孵育8min后,轻弹混匀或者轻微涡旋混匀,即可获得检测样品。
实施例4
本实施例与实施例3的区别在于:步骤(5)中,将tebuffer改用为ddh2o,即:在步骤(4)得到的所述沉淀中加入新鲜配制的75%无水乙醇洗涤两次,弃掉上清液,将沉淀室温晾干5min;加入20μlddh2o,室温孵育8min后,轻弹混匀或者轻微涡旋混匀,即可获得检测样品。
对比例1
本实施例提供了一种液态奶中微生物总dna的提取方法,所述提取方法包括以下步骤:
(1)样品处理:室温均质牛奶样品,取1.0ml牛奶样品室温13000xg离心5min,沉淀样品,弃掉上清液,得到沉淀物;
(2)细胞裂解:在步骤(1)得到的所述沉淀物中加入500μl的enzymaticlysisbuffer和1μllysozyme,上下吹打混匀,37℃孵育1h,后,加入3μl蛋白质k到裂解液中,55℃热孵育1h,得到重悬浮液,将所述重悬浮液转移到一个含有0.8g玻璃珠子的离心管中振荡10min,得到裂解液,室温条件下,13000xg离心5min,避开沉淀及玻璃珠,取上清液到一个新离心管中;
(3)纯化dna:向步骤(2)得到的所述上清液中加入1倍体积的酚氯仿进行抽提,旋涡振荡,直至白色悬浊液出现,室温孵育5min,然后离心取上清液;所述上清液中加入1倍体积的酚氯仿进行抽提,旋涡振荡,直至白色悬浊液出现,室温孵育5min,然后在4℃下,16000xg离心10min,取上清液;
(4)回收dna:在步骤(3)得到的所述上清液中加入1μl的glycogen、1/10体积的3m醋酸钠和1倍体积异丙醇,所述醋酸钠的ph为5.2,上下颠倒混匀5-6次,-20℃孵育30min,4℃,16000xg离心15min,弃掉上清液,并同时保持沉淀在离心管中;
(5)溶解dna:在步骤(4)得到的所述沉淀中加入新鲜配制的75%无水乙醇洗涤两次,弃掉上清液,将沉淀室温晾干3-5min;加入20μltebuffer,室温孵育8min后,轻弹混匀或者轻微涡旋混匀,即可获得检测样品。
对比例1与本发明实施例3的区别在于:实施例3直接在含有玻璃珠的裂解液中加入1倍体积的酚氯仿进行抽提,对比例1经过离心取上清液,然后在上清液中加入1倍体积的酚氯仿进行抽提。
不同提取方法得到的宏基因组dna样品的1%琼脂糖凝胶电泳对比如图1所示,3-4为本发明实施例3得到的检测样品,3-3为对比例1得到的检测样品,marker条带分别为100bp,250bp,500bp,750bp,1000bp,2000bp,3000bp,5000bp,8000bp。
从图1中可以看出,本发明实施例3得到的基因组dna条带更加明亮,清晰,完整。说明,sds裂解后,直接进行有机试剂抽提,不仅可以减少浪费、节省时间,还可以减少dna损失。
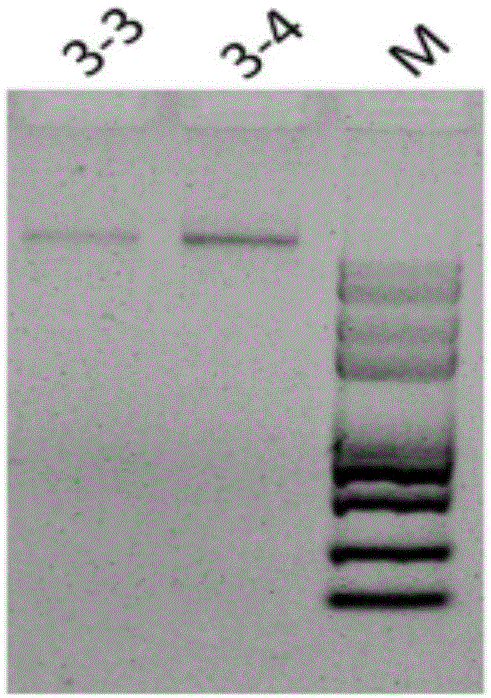
对实施例3和实施例4得到的宏基因组dna样品分别进行16srdna扩增子测试
(1)本发明中16srrna扩增分两次进行,第一次扩增包括依次进行的初步扩增和进一步扩增,引物采用16sv3forwardprimer和16sv4reverseprimer;第二次扩增的引物为forward和reverse。第一次扩增先用短链引物进行扩增以降低非特异性扩增,再采用长链扩增以达到富集和添加测序接头的目的。
①第一次扩增体系如下:
16sv3forward的序列为:
5'-cctacgggnggcwgcag-3';
16sv4reverse的序列为:
5'-gactachvgggtatctaatcc-3'。
扩增条件:
初步扩增过程为5个循环;
进一步扩增包括:变性95℃,20s
退火60℃,30s
延伸72℃,30s进一步扩增过程为20个循环;
终延伸72℃,5min
保温4℃
②第二次扩增体系如下:
移液器吹打多次混合均匀,避免较多气泡产生;
forward的序列为:
5'-tcgtcggcagcgtcagatgtgtataagagacag--cctacgggnggcwgcag-3';
reverse的序列为:
5'-gtctcgtgggctcggagatgtgtataagagacag--gactachvgggtatctaatcc-3'。
扩增条件:
预变性98℃,30s
变性98℃,10s
退火/延伸65℃,75s
扩增过程为8个循环;
终延伸65℃,5min
保温4℃
③扩增产物检测
16srdna扩增结束后,取3μl样品进行琼脂糖凝胶电泳检测,dna建库起始量为500ng,结果如图2所示。图2中,m为dna分子量标准,m条带分别为100bp,250bp,500bp,750bp,1000bp,2000bp,3000bp,5000bp,8000bp;3-2为实施例3得到的宏基因组dna样品,3-3为实施例4得到的宏基因组dna样品。从图2中可以看出,3-2和3-3扩增较好,基本没有非特异性扩增出现;实验结果也可以反映出ddh2o(双蒸水)中无微生物干扰。
④扩增产物回收及质控
从胶图上看到,非特异性扩增的量比较少,与主带大小相差较大,所以选择使用磁珠两步纯化的方法进行回收目的dna片段,具体步骤如下:
(1)在pcr产物中补加ddh2o,定容至100μl,并移液器吹打混匀;
(2)加入55μl平衡室温后磁珠到上述pcr管中,移液器吹打多次混匀,混合液颜色均匀为止;室温孵育5分钟;
(3)把pcr管放到磁力架上,静置3-5分钟,待液体澄清后,转移上清液到一个新的离心管中,小心不要吸到磁珠;加入45μl磁珠到上清液中,吹打多次混匀后,室温孵育5分钟;孵育结束后,把离心管放到磁力架上,静置3-5分钟,待上清液澄清后,弃掉上清液;
(4)保持离心管在磁力架上,加入200μl新鲜配制的80%乙醇,上下颠倒数次,静置30s,弃掉上清;重复一次上述过程后,稍微离心,尽可能吸掉残留液体;保持离心管在磁力架上,室温晾干3-5min,即磁珠表面没有液滴残留或者出现轻微裂痕;晾干后,加入40μl10mmtris-hcl(ph8.0),吹打混匀后,室温孵育5min;孵育结束后,把离心管放到磁力架上,待液体澄清后,吸取上清液到一个新离心管中,并做好标记;
对纯化后产物进行质量控制,分别为qubit定量和agilent2100定性,qubit定量结果如表1:
表1qubit检测结果
实施例3得到的基因组dna的agilent2100定性检测结果如图3所示,实施例4得到的基因组dna的agilent2100定性检测结果如图4所示。
从图3和图4中可看出,扩增条带单一,无较多的背景杂峰,大小基本一致,符合16sv3v4区域dna片段大小。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!