DNA零件,途径和基因组的进化指导的多重DNA装配的制作方法
在过去十年中,高通量dna测序已经改变了生物科学和医学的每个方面。今天,我们处在一个新时代的开端,生物科学从一个以知识为导向的学科转变为复杂生物系统的应用相关工程,从而使其高度创新的技术潜力倍增和资本化,以生产出应用于医学、农业、材料科学和可持续食物和生物能源生产的多种分子。
低成本从头dna合成技术的最新进展现在首次提供了通过编写长dna分子来编程生物功能的能力。未来,dna的从头合成将对生物学和医学产生比基因组测序革命更大的变革性影响。在这种转变期间,新的使能技术,如本文提出的进化指导的多重基因组装配过程,将是成本和时间有效地制造合成dna设计以加速复杂生物系统的生物工程的关键。
尽管最近技术在从头dna合成能力、染色体装配和编辑工具方面突破,快节奏的从头dna合成仍然是有效制造具有完全确定的基因组成的平台生物的合成生物学的主要限速步骤。
用于从头dna合成的基于硅和芯片的方法现在能够大规模制造短双链dna序列(如通过使用twist公司、gen9公司、赛默飞世尔公司(thermo-fisher)技术所例证的)。这些方法能够同时产生长千上万个短寡核苷酸,这些短寡核苷酸装配成1kb长的双链dna分子,并且在下一次迭代中随后连接成更高级的装配。然而,由于固相化学的微型化和限制,先进的低成本寡核苷酸制造技术不能保证每个dna嵌段都能以流水线方式制造。
目前大规模dna制造工艺的基石仍遵循经典化学合成采用的设计原则:首先定义所希望的dna分子的序列,并且然后在一系列连续的化学反应后构建精确的拷贝。在从头dna合成期间,dna分子(设计)的结构(碱基对序列)保持恒定。在随后的分离和测序过程期间丢弃与初始序列设计(或其部分)不同一的副产物或中间体。寡核酸合成和聚合酶链装配(pga)反应期间的合成错误需要重复、优化和改进反应条件,直到达到足够的产率以进行随后的高级dna装配步骤。由于内在的分层性质,该过程严格依赖于从前面的装配水平成功制造每个单独的结构单元。因此,合成途径、基因簇和由成百上千个dna嵌段组成的整个基因组的工程化很快成为不可克服的问题,因为即使一个单一的缺失的dna嵌段也阻碍了分层装配,并且因此阻止了dna设计的完成。因此,当前的基因组制造被推迟,直到在从头dna合成尝试的迭代循环期间已经获得了每个难以合成的dna嵌段。
技术实现要素:
基于上述背景,本发明的目的是提供产生可包含完整途径、基因簇或整个基因组的大dna构建体的方法。
该目的通过具有根据权利要求1所述的特征的方法实现。优选的实施例陈述于从属权利要求和以下说明中。
据此,本发明的第一方面涉及用于制造目的大dna构建体的方法。该方法包括以下步骤:
-提供包含多个遗传元件的计算机模拟模板dna构建体;
-使计算机模拟模板dna构建体经历计算优化步骤,其中通过中性序列改变,特别是通过在包含在一个或多个蛋白质编码序列内的情况下中性密码子替换或通过包含在一个或多个基因间序列内的情况下中性碱基取代插入、缺失或同义序列替换,从计算机模拟dna构建体模板中去除抑制从头dna合成的一个或多个序列,产生优化的计算机模拟dna构建体,并且起始密码子不被去除或替换;
-在分配步骤中将优化的计算机模拟dna构建体分配成多个原始计算机模拟装配单元,其中优化的计算机模拟dna构建体的分配使得多个原始计算机模拟装配单元中的每两个相邻单元共享末端同源区,其中每个末端同源区不同于任何其他末端同源区;
-使多个原始计算机模拟装配单元中的每个单元经历计算同义序列重编码步骤,其中
·通过中性序列改变为多个原始计算机模拟装配单元中的每个单元产生一个或多个同义计算机模拟装配单元,并且没有改变末端同源区或起始密码子,并且
·产生包含多个计算机模拟原始装配单元的单元以及一个或多个同义计算机模拟装配单元的计算机模拟装配变体池,从而产生计算机模拟变体池文库;
-从头合成所述计算机模拟变体池文库的每个计算机模拟装配变体池中的一个或多个单元,从而产生核酸装配单元文库;以及
-在装配步骤中将核酸装配单元文库在体外或体内装配成目的dna构建体。
无论何时构建体或装配单元被称为“计算机模拟”,在本说明书的上下文中应理解,相应的构建体或装配单元以数字序列的形式存在,例如,以计算机可读格式编码。
特别地,每当两个相邻的装配单元共享末端同源区时,应理解,两个相邻的装配单元包含相应的末端同源区,两个装配单元装配在该末端同源区上。
在本说明书的上下文中,术语“中性序列改变”特别地是指序列的改变,这种改变不影响相应序列的生物学功能,例如,仅引起沉默突变。
中性序列改变的非限制性实例包括
-蛋白质编码序列中的中性密码子替换,和
-基因间序列中的中性碱基取代、插入或缺失或同义序列替换。
在本说明书的上下文中,术语“基因间序列”特别地指位于两个基因之间的dna的非编码段。
在本说明书的上下文中,术语“中性密码子替换”是指在目的dna构建体的蛋白质编码序列内或在计算机模拟装配单元内将密码子用编码相同氨基酸残基的不同密码子更换。
在本说明书的上下文中,术语“同义序列替换”特别地是指将在模板计算机模拟模板内的一个或多个基因间序列用一个或多个提供相似生物学功能的序列替换。
在本说明书的上下文中,术语“中性碱基取代、插入或缺失”特别地是指不影响相应序列的生物学功能的碱基取代、插入或缺失。
特别地,通过用一个或多个不抑制从头合成,特别是编码相同多肽或提供相似生物学功能的同义序列替换抑制从头dna合成的一个或多个序列来将其去除,其中所述一个或多个同义序列是通过中性序列改变产生的,例如,蛋白质编码序列中的中性密码子替换或基因间序列内的中性碱基取代、插入或缺失或同义序列替换。
本领域技术人员理解,上述原始计算机模拟装配单元中的每一个以及因此除了初始和末端装配单元之外的一个或多个同义计算机模拟装配单元中的每一个包括两个同源区,相应的装配单元可以在其上与前面的装配单元和随后的装配单元装配。
抑制从头dna合成的序列的非限制性实例包括具有高gc含量(特别地是高于50%)的序列、具有6bp或更长的长度的均聚序列、二核苷酸和三核苷酸重复、直接重复和更长的发夹(特别是具有在8bp至12bp或更长的范围内的长度)。
体外装配的非限制性实例是吉普森(gibson)装配,其中该核酸装配单元装配在末端同源区上。用于体内装配的非限制性实例是酵母装配,其中用核酸装配单元转化酵母细胞,特别是通过合适的媒介物如载体转化,并且核酸装配单元在酵母细胞内装配。
有利地,本发明的方法克服了关于装配单元的已知方法的限制,这些方法通过提供一个或多个同义装配单元几乎不可或甚至完全不可合成,通过这种方式用于成功装配的所有所需装配单元的成功从头合成的可能性大大增加。
此外,本发明的方法不仅允许产生大dna构建体,在计算优化步骤和/或计算同义序列重编码步骤中通过非中性密码子或非同义序列替换也可能产生其变体。
因此,本发明的第二方面涉及用于制造目的dna构建体变体的方法,包括以下步骤:
-提供包含多个遗传元件的原始计算机模拟dna构建体;
-使原始的计算机模拟dna构建体经历计算优化步骤,其中通过中性序列改变从计算机模拟dna构建体模板中去除抑制从头dna合成的一个或多个序列,产生优化的计算机模拟dna构建体,并且起始密码子未被去除或替换;
-在分配步骤中将优化的计算机模拟dna构建体分配成多个原始计算机模拟装配单元,其中优化的计算机模拟dna构建体的分配使得多个原始计算机模拟装配单元中的每两个相邻单元共享末端同源区,其中每个末端同源区不同于任何其他末端同源区;
-使多个原始计算机模拟装配单元中的每个单元经历计算突变序列重编码步骤或计算同义序列重编码步骤,其中
·在计算突变序列重编码步骤中,通过非中性序列改变,为多个原始计算机装配单元中的一个或多个单元产生一个或多个突变体计算机模拟装配单元,并且不改变末端同源区或起始密码子,并且产生包含一个或多个突变体计算机模拟装配单元的计算机模拟装配突变体池,从而产生计算机模拟突变体池的相应文库;以及
·在计算同义重编码步骤中,通过中性序列改变为不进行计算突变序列重编码步骤的多个原始计算机模拟装配单元中的每个单元产生一个或多个同义计算机模拟装配单元,并且没有末端同源区或起始密码子被改变,并产生包含多个原始计算机模拟装配单元和一个或多个同义计算机模拟装配单元的单元的计算机模拟装配变体池,从而产生计算机模拟变体池的相应文库;
-从头合成计算机模拟变体池文库的每个计算机模拟装配变体池的一个或多个单元和计算机模拟突变体池文库的每个计算机模拟突变体池的一个或多个单元,从而产生核酸装配单元文库;以及
-在装配步骤中将核酸装配单元文库在体外或体内装配成目的dna构建体变体。
在本说明书的上下文中,术语“非中性序列改变”特别地是指序列的改变,这种改变影响相应序列的生物学功能。
非中性序列改变的非限制性实例包括
-蛋白质编码序列中的非中性密码子替换,
-通过蛋白质编码序列中的开放阅读框内的碱基插入或缺失引入读框移位,和
-基因间序列中的非中性碱基取代、插入、非同义序列替换。
在本说明书的上下文中术语“非中性密码子替换”是指在目的dna构建体的蛋白质编码序列内或在计算机模拟装配单元内将密码子用编码不同氨基酸残基的不同密码子更换。
在本说明书的上下文中,术语“非同义序列替换”特别地是指将在模板计算机模拟模板内或在不提供相似生物学功能的计算机模拟装配单元内替换一个或多个基因间序列。
在本说明书的上下文中,术语“非中性碱基取代、插入或缺失”特别地是指影响相应序列的生物学功能的碱基取代、插入或缺失。
在某些实施例中,在计算优化步骤中通过中性密码子替换,从计算机模拟dna构建体模板中去除包含在一个或多个蛋白质编码序列内并抑制从头dna合成的一个或多个序列。
在某些实施例中,在计算优化步骤中通过中性碱基取代、插入或缺失或同义序列替换,从计算机模拟dna构建体模板中去除包含在一个或多个基因间序列内并抑制从头dna合成的一个或多个序列。
用于制造目的dna构建体变体的可替代方法包括以下步骤:
-提供包含多个遗传元件的原始计算机模拟dna构建体;
-使原始的计算机模拟dna构建体经历计算诱变步骤,其中通过非中性序列改变从原始计算机模拟dna构建体中去除抑制从头dna合成的一个或多个序列,产生优化的突变体计算机模拟dna构建体,并且起始密码子未被去除或替换;
-在分配步骤中将优化的突变体计算机模拟dna构建体分配成多个原始计算机模拟装配单元,其中优化的突变体计算机模拟dna构建体的分配使得多个原始计算机模拟装配单元中的每两个相邻单元共享末端同源区,其中每个末端同源区不同于任何其他末端同源区;
-使多个原始计算机模拟装配单元中的每个单元经历计算同义序列重编码步骤,其中
·通过中性序列改变为多个原始计算机模拟装配单元中的每个单元产生一个或多个同义计算机模拟装配单元,并且没有改变末端同源区或起始密码子,并且
·产生包含多个原始计算机模拟装配单元的单元以及一个或多个同义计算机模拟装配单元的计算机模拟装配变体池,从而产生计算机模拟变体池的相应文库;
-从头合成计算机模拟变体池文库的每个计算机模拟装配变体池中的一个或多个单元,从而产生核酸装配单元文库;以及
-在装配步骤中将核酸装配单元文库在体外或体内装配成目的dna构建体变体。
在某些实施例中,在计算诱变步骤中通过一个或多个蛋白质编码序列内的非中性密码子替换或碱基缺失,从原始计算机模拟dna构建体中去除包含在一个或多个蛋白质编码序列内并抑制从头dna合成的一个或多个序列。
在某些实施例中,在计算诱变步骤中通过非中性碱基取代、插入或缺失或通过非同义替换从原始计算机模拟dna构建体中去除包含在一个或多个基因间序列内并抑制从头dna合成的一个或多个序列。
可替代地,这种变体可以通过非中性序列改变如非中性密码子替换或用原始dna构建体的非同义序列替换以计算机模拟的方式产生,产生计算机模拟突变体dna构建体,然后进行根据本发明上述方面的方法,得到相应核酸形式的突变dna构建体。
因此,用于制造目的dna构建体变体的另外的可替代方法包括以下步骤:
-提供包含多个遗传元件的计算机模拟dna构建体模板;
-使计算机模拟dna构建体模板经历计算诱变步骤,其中计算机模拟模板dna构建体内的一个或多个序列通过非中性序列改变而改变,产生突变体计算机模拟dna构建体
-使突变体计算机模拟dna构建体经历计算优化步骤,其中通过中性序列改变从计算机模拟dna构建体模板中去除抑制从头dna合成的一个或多个序列,产生优化的突变体计算机模拟dna构建体,并且起始密码子未被去除或替换;
-在分配步骤中将优化的突变体计算机模拟dna构建体分配成多个原始计算机模拟装配单元,其中优化的突变体计算机模拟dna构建体的分配使得多个原始计算机模拟装配单元中的每两个相邻单元共享末端同源区,其中每个末端同源区不同于任何其他末端同源区;
-使多个原始计算机模拟装配单元中的每个单元经历计算同义序列重编码步骤,其中
·通过中性序列改变为多个原始计算机模拟装配单元中的每个单元产生一个或多个同义计算机模拟装配单元,并且没有改变末端同源区或起始密码子,并且
·产生包含所述多个原始装配单元以及一个或多个同义计算机模拟装配单元的所述单元的计算机模拟装配变体池,从而产生计算机模拟变体池文库
-从头合成计算机模拟变体池文库的每个计算机模拟装配变体池中的一个或多个单元,从而产生核酸装配单元文库;以及
-在装配步骤中将核酸装配单元文库在体外或体内装配成目的dna构建体变体。
在某些实施例中,在计算诱变步骤中通过一个或多个蛋白质编码序列内的非中性密码子替换或碱基缺失,包含在一个或多个蛋白质编码序列内的一个或多个序列被改变。
在某些实施例中,在计算诱变步骤中通过非中性碱基取代、插入或缺失或通过非同义序列替换,包含在一个或多个基因间序列内的一个或多个序列被改变。
在某些实施例中,从计算机模拟dna构建体模板中去除cg含量等于或高于50%、60%、70%、80%或85%并且长度在21个碱基对至99个碱基对范围内的序列。在某些实施例中,从计算机模拟dna构建体模板中去除cg含量等于或高于70%且长度为21个碱基对的序列。在某些实施例中,从计算机模拟dna构建体模板中去除cg含量等于或高于85%且长度为99个碱基对的序列。
在某些实施例中,核酸装配单元文库在装配步骤之前在扩增步骤中扩增,产生扩增的核酸装配单元文库,其中该扩增的核酸装配单元文库在装配步骤中装配成目的dna构建体或其变体。
在某些实施例中,每个计算机模拟装配单元变体或突变体池的一个或多个单元合成为双链dna,其中特别地是双链dna附接至固体支持物或存在于溶液中。
在某些实施例中,将第一可分离的适配子序列添加到每个计算机模拟装配变体或突变体池的每个单元的一个末端,并且将第二可分离的适配子序列添加到每个计算机模拟装配变体或突变体池的每个单元的另一个末端,其中
-第一可分离的适配子序列和第二可分离的适配子序列具有不同的序列,并且其中任选地,在扩增步骤中使用能够退火至第一可分离的适配子序列的第一引物和能够退火至第二可分离的适配子序列的第二引物,并且
-在装配步骤之前,从核酸装配单元文库的每个单元或扩增的核酸装配单元文库中去除第一可分离的适配子序列和第二可分离的适配子序列。
技术人员理解,添加到计算机模拟装配单元中的第一可分离的适配子序列和第二可分离的适配子序列被合成为附接至相应核酸装配单元的核酸序列。
在某些实施例中,第一可分离的适配子序列包含第一引物结合区和第一切割位点,其中第一切割位点在第一引物结合区和每个计算机模拟装配变体或突变体池的每个单元的一个末端之间安排。
在某些实施例中,第二可分离的适配子序列包含第二引物结合区和第二切割位点,其中第二切割位点在第二引物结合区和每个计算机模拟装配变体或突变体池的每个单元的另一个末端之间安排
在某些实施例中,第一切割位点和第二切割位点可被不同的内切核酸酶特异性识别。
在某些实施例中,第一引物包含与第一引物结合区至少80%、85%、90%、95%、99%或100%相同或互补的核酸序列或由其组成。在某些实施例中,第二引物包含与第二引物结合区至少80%、85%、90%、95%、99%或100%相同或互补的核酸序列或由其组成。
在某些实施例中,目的dna构建体或其变体是线性核酸分子、环状核酸分子如质粒或人工染色体。
在某些实施例中,目的dna构建体具有至少10,000个碱基对的长度。在某些实施例中,目的dna构建体具有至少1000,000个碱基对的长度。
在某些实施例中,多个原始计算机模拟装配单元中的每个单元彼此独立地具有500个碱基对至3,000个碱基对范围内的长度。
在某些实施例中,彼此独立的每个末端同源区具有15个碱基对至35个碱基对的长度。
在某些实施例中,遗传元件选自操纵子、启动子、开放阅读框、增强子、沉默子、外显子、内含子或基因。
在某些实施例中,目的dna构建体、原始计算机dna模拟构建体或计算机模拟dna构建体模板包含一个或多个基因簇或全基因组或由其组成。在某些实施例中,目的dna构建体、原始计算机dna模拟构建体或计算机模拟dna构建体模板包含对应于一个或多个代谢途径的多种遗传元件。
在某些实施例中,模板dna构建体或原始dna构建体是天然存在的或人工的。
这种人工dna构建体可以源自天然存在的核酸如基因簇或基因组,其中并入一个或多个外源遗传元件如基因、启动子、操纵子或开放阅读框,和/或天然存在的遗传元件已被替换和/或缺失。这种人工dna构建体也可以是源自多个不同生物的多个遗传元件的嵌合体。
在某些实施例中,计算机模拟dna构建体模板是天然或人工来源的功能性dna构建体的变体,特别地意指由功能性遗传元件构成的dna构建体,其中通过碱基或序列的插入或缺失、或序列的倒置或非中性密码子替换而使一个或多个遗传元件变得无功能。
在某些实施例中,末端同源区包含在蛋白质编码序列内,其中所述末端同源区与蛋白质编码序列同框开始。在某些实施例中,末端同源区包含在基因间序列内。
在某些实施例中,分配步骤包括
-将优化的计算机模拟dna构建体或优化的突变体计算机模拟dna构建体分配成多个计算机模拟区段装配单元,其中每两个相邻的计算机模拟区段装配单元共享区段末端同源区,其中特别地是一个区段末端同源区与任何其他的不同;
-将多个计算机模拟区段装配单元中的每个区段分配成多个计算机模拟嵌段装配单元,其中每两个相邻的嵌段装配单元共享嵌段末端同源区,其中特别地是一个嵌段末端同源区与任何其他的不同;以及
-将多个计算机模拟嵌段装配单元中的每个单元分配成多个计算机模拟子嵌段装配单元,其中每两个相邻的子嵌段装配单元共享子嵌段末端同源区,从而产生如以上所描述的多个原始计算机模拟装配单元。
在某些实施例中,装配步骤包括
-将核酸装配单元文库或对应于计算机模拟嵌段装配单元的扩增的核酸装配单元文库的单元分别地汇集和装配成核酸嵌段装配单元,产生多个核酸嵌段装配单元;
-将对应于计算机模拟区段装配单元的核酸嵌段装配单元分别地汇集和装配成核酸区段装配单元,产生多个核酸区段装配单元;以及
-将核酸区段装配单元汇集和装配成目的dna构建体或其变体。
在某些实施例中,第一可分离的适配子序列是或包含区段适配子序列,并且第二可分离的适配子序列是或包含嵌段适配子序列,其中
-对应于相同的计算机模拟区段装配单元的每个计算机模拟装配变体或突变体池的单元具有相同的区段适配子序列,
-对应于相同的计算机模拟嵌段装配单元的每个计算机模拟装配变体或突变体池的单元具有相同的嵌段适配子序列,
-每个区段适配子序列彼此不同,并且
-每个嵌段适配子序列彼此不同
在某些实施例中,将区段适配子序列添加到相应的计算机模拟装配变体或突变体池的相应单元的5'末端,并将嵌段适配子序列添加到相应单元的3'末端。
在某些实施例中,多个计算机模拟区段装配单元中的每个单元彼此独立地具有10,000个碱基对至50,000个碱基对范围内的长度。
在某些实施例中,多个计算机模拟嵌段装配单元中的每个单元彼此独立地具有2,000个碱基对至10,000个碱基对范围内的长度。
在某些实施例中,区段末端同源区中的每个彼此独立地具有35个碱基对至200个碱基对范围内的长度。
在某些实施例中,嵌段末端同源区中的每个彼此独立地具有35个碱基对至90个碱基对范围内的长度。
通过以下某些实施例的详细描述、实例和附图进一步阐述本发明,从中可以得出进一步的实施例和优点。这些实例意指阐述本发明但不是限制其范围。
附图说明
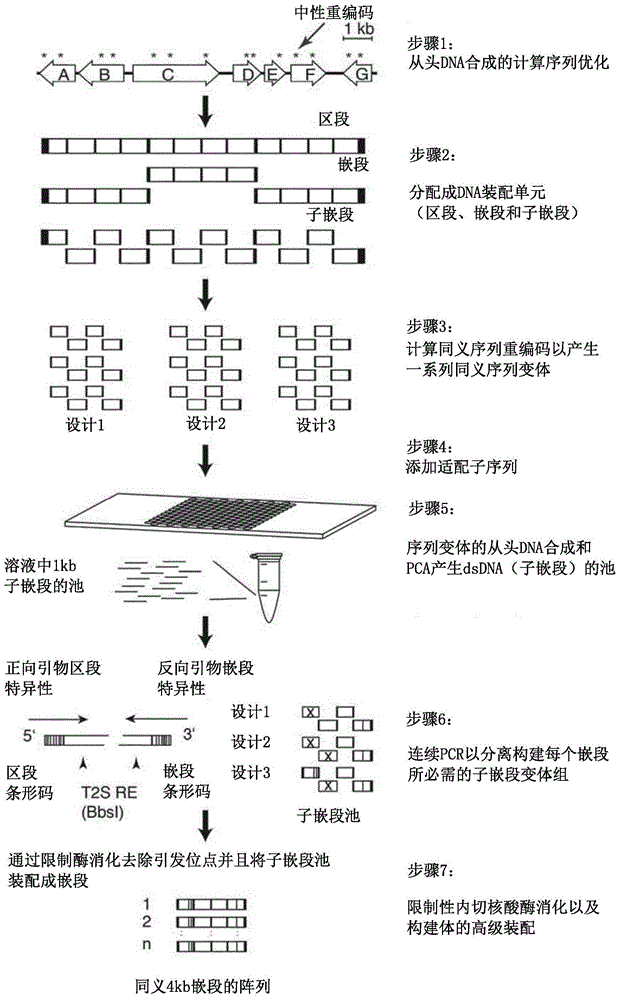
图1示出了进化指导的多重基因组装配过程的工作流程。
图2示出了773'851碱基对长驯化基因组设计的图谱,以及分配设计指示通过现有方法的合成成功率。
图3示出了子嵌段的多重dna装配成嵌段(a)分配设计的概述。(b)子嵌段设计变体的从头dna合成产率的概述。将条形码化的子嵌段进行pcr扩增,并在1%琼脂糖凝胶上分离。对于设计1:sb8,sb12;设计2:sb5、sb12、sb13;设计3:sb4、sb9、sb13从头dna合成失败,(c)通过条形码特异性pcr扩增产生的用于嵌段装配的子嵌段的池。每个pcr反应产物含有已经由pca成功合成的特定嵌段装配的所有子嵌段设计变体的组。(d)区段25的多重嵌段装配反应。通过pcr跨子嵌段连接确认对所有5个嵌段的正确装配。每个嵌段由4个名为a-d的子嵌段装配而成,并且通过pcr测试的连接相应地标记为ab、bc、cd。(e)使用限制性消化从克隆载体中释放嵌段。较低的带对应于4kb大小的嵌段片段。(f)将嵌段装配成放大嵌段连接的20kb区段的pcr验证。(g)在产生pseg25的目标载体pmr10y中装配的区段25构建体的大小的验证。将超螺旋质粒pseg25的大小与携带19kb插入物的超螺旋参考质粒pmr10y(白色箭头)进行比较。
表1以236个-4kb嵌段分配的驯化基因组的dna合成产率。
表2子嵌段设计变体之间的碱基取代率。
表3来自区段25的子嵌段设计变体的从头dna合成产率。
表4使用子嵌段变体池的嵌段装配反应的效率。
表5用于分配的适配子序列。
表6子池pcr扩增使用的条形码引物。
表7用于嵌段装配的pcr验证的引物。
表8菌株列表。
具体实施方式
本发明实现了利用从头dna合成以及工程化至基因组规模,从而通过称为基因组的进化指导的多重dna装配的可扩展的dna合成过程减少了生物系统设计的时间和成本。该方法解决了从许多小的双链dna嵌段以分层方式制造大规模dna构建体的问题,每个dna嵌段都不能以100%的成功率产生。
进化指导的多重dna装配不是构建单个dna序列设计,而是平行采用多个同义dna序列变体,并以组合装配方式选择具有最佳合成和装配可行性的那些序列变体。
在某些实施例中,本发明的多重基因组装配方法基于7步法(图1)。该方法的主要阶段是i)用于从头dna合成的dna设计(上述称为目的dna构建体)的计算优化,ii)分配成dna装配单元(区段、嵌段和子嵌段,上述称为原始计算机模拟装配单元),iii)计算同义序列重编码以产生一系列同义序列变体(上述称为同义计算机模拟装配单元),iv)向子嵌段设计变体添加适配子序列,v)同义序列变体池的从头dna合成,vi)连续pcr以分离构建每个嵌段所必需的子嵌段变体组,vii)去除末端pcr条形码序列以及构建体的高级装配。
本发明的关键原则是dna设计是序列优化的并且分配成用作高级dna装配的冗余装配单元的同义变体。因此,dna合成并不严重依赖于所有构建单元的成功合成
第一步:用于从头dna合成的设计的计算优化-优化dna序列设计(大小高达整个人工基因组)用于从头dna合成,以产生合成优化的dna设计。
在某些实施例中,dna序列设计代表核酸分子、质粒或一个或多个人工染色体。
在某些实施例中,dna序列设计包含多于(>)10.000bp,特别地是>1.000.000bp。
使用基因组书法家(genomecalligrapher)软件算法或相似的计算算法,通过中性重编码(同义密码子替换)重构所述dna序列设计的蛋白质编码序列,以消除已知抑制从头dna合成的不允许的序列模式。序列设计和序列重构的方法描述于ep15195390.8中,其通过引用以其整体并入本文。用于通过中性重编码、密码子优化及其使用方法进行dna重构的基因组书法家软件算法描述于(christen,m.,deutsch,s.,&christen,b.(2015).genomecalligrapher:awebtoolforrefactoringbacterialgenomesequencesfordenovodnasynthesis[基因组书法家:一种用于重构细菌基因组序列的网络工具,用于从头dna合成].acssyntheticbiology[acs合成生物学],4(8),927-934.http://doi.org/10.1021/acssynbio.5b00087),通过引用以其整体并入本文。
第二步:分配为dna装配单元-合成优化的dna设计被分配为用于分层装配的dna单元(区段、嵌段、子嵌段)。可整合多达三个装配水平。在第一水平,子嵌段组被装配成嵌段。在第二装配水平嵌段组进一步装配成区段,最终装配成最终的大规模dna构建体。随着装配水平的提高,dna装配单元的大小增加,并且理想地,对于子嵌段是在500-3'000bp范围内,对于嵌段是在2'000-10'000bp范围内,以及对于区段是在10'000-50'000bp范围内。在整个分配设计中,在相邻装配单元之间限定短末端同源区(thr)(大小为从15至200bp)。这些区提供用于本领域已知的高级装配以及用于将相邻的装配单元连接成高级构建体的末端序列同源性。thr的边界根据以下设计规则定义:
-所述thr的边界位于基因间序列内或蛋白质编码dna序列(cds)内。
-如果thr属于cds,则每个装配单元的边界设定为框内。
本发明的一个方面涉及用于分配大的数千碱基dna序列的计算方法,其中软件算法(基因组分配器)用于实现dna序列分配为分层装配水平并根据上述指定的设计规则定义末端同源区。所述算法中整合了三个装配水平:
dna序列分配算法使用带注释的dna序列文件(genbank文件)作为输入,并且包括以下步骤:
a)将dna序列分配为具有使用者定义的区段大小的dna区段(理想地在10至100kb的范围内,大小偏差小于10%,包括区段thr)。每个区段与之前(5')区段(理想地在35-200bp的范围内)共享末端同源区。调整落入编码序列内的thr的边界以适合相应的阅读框。这种调整在thr的产生和优化期间完成,dna区段携带覆盖与目的载体同源的相邻5'和3'末端适配子序列,并且任选地含有用于在酵母中限制性内切核酸酶消化、克隆或高级装配的接头序列。区段的序列记录(包括末端适配子)写在fasta文件中,并且没有适配子的区段边界在genbank输出文件中注释。在某些实施例中,根据相似于以下(b)中指定的嵌段水平的thr设计规则的thr设计规则来优化区段水平的thr。
b)使用以下设计规则将dna区段进一步细分为dna嵌段:dna嵌段具有使用者定义的大小(理想地在2至10kb的范围内,包括区段和嵌段thr和适配子序列的长度,并且具有均匀的大小,大小偏差小于10%)。dna嵌段通过使用者定义的嵌段thr(理想地在35至90bp的范围内)与相邻嵌段重叠。调整嵌段thr的边界以适合相应的读取框。dna嵌段携带覆盖与i)目的载体同源性的相邻适配子序列和ii)任选地含有用于限制性内切核酸酶消化和克隆到目的载体的接头序列
c)分析在嵌段水平的装配单元的末端同源区是否存在干扰本领域已知的同源末端连接的序列特征,并且用于连接相邻的装配单元。通过将thr上游或下游移位至不再包括重复序列或任何另外的重复序列(非唯一序列模式)并重新调整相应的嵌段边界,去除thr内的重复大小大于使用者指定的限制(8bp)的发夹和直接重复序列。计算在每个区段的dna嵌段的thr区内多次出现的相同子链(即非唯一序列)。通过产生具有不再包括有问题的非唯一序列模式的移位thr的一组分配变体来鉴定并去除在嵌段水平的多个thr内发生的最大相同子链。针对多个thr内重复、发夹的出现和子链的多次出现迭代地评估这些分配设计变体。然后使用量度来鉴定最佳分配设计变体,该变体i)显示不存在重复,以及ii)不存在非唯一序列,以及iii)需要thr区的最少再定位。此外,为了优化thr,不允许嵌段大小偏离使用者提供的平均嵌段大小超过10%。选择最佳分配设计并调整相应的嵌段边界。重复thr优化,直到达到使用者为相同子链定义的大小下限(8bp)。调整蛋白质编码序列内的thr以适合两个末端的相应阅读框。在每个嵌段的thr的创建和优化期间完成该调整。在完成dna嵌段分配后,嵌段的序列记录被写入fasta文件(包括适配子)中,并且嵌段边界在genbank输出文件中注释(没有嵌段适配子)。
d)使用以下设计规则将dna嵌段进一步细分为dna子嵌段:dna子嵌段通过使用者定义的thr(理想地在15至35bp的范围内)与相邻子嵌段重叠。没有子嵌段偏离超过使用者定义的最大子嵌段大小(理想地在500至3000bp的范围内,包括区段、嵌段和子嵌段thr以及相应的适配子序列的长度)10%。
e)dna子嵌段的末端同源区根据如c)中针对在嵌段水平的thr优化采用的相同途径进行优化。dna子嵌段携带覆盖与目的载体同源性的相邻适配子序列和任选地含有用于限制性内切核酸酶消化用于亚克隆的接头序列。子嵌段写入fasta文件(包括子亚克隆适配子和pcr适配子),并且子嵌段在genbank输出文件中注释(没有子嵌段适配子)。
在某些实施例中,5'和3'适配子序列含有特异性引物退火位点,其允许用于高级装配的dna单元组的平行pcr扩增。
在某些实施例中,如果缝合寡核苷酸用于随后的dna单元装配,则可以省略5'和3'适配子序列。
第三步:计算同义序列重编码以产生一系列同义序列变体-分配优化的dna设计是序列被重编码以产生一组(n)个同义序列变体。因此,蛋白质编码序列内的密码子被同义密码子取代。在某些实施例中,在引入碱基取代、插入或缺失或用覆盖相似生物学功能的同义序列替换基因间序列后产生基因间序列内的变体。已经为装配过程分配thr的区被排除在重编码之外并且依序保持不变。每个蛋白质编码序列内的多肽序列信息由一系列用于20个氨基酸的61个核苷酸三联体编码。遗传密码的这种冗余允许特定密码子被仍然编码相同氨基酸的同义密码子替换。通过重编码的过程,产生一组编码相同的蛋白质但核苷酸序列不同的序列变体。用于dna重构、密码子优化及其使用方法的基因组书法家软件算法描述于(christen,m.,等人http://dos.org/10.1021/acssynbio.5b00087),通过引用以其整体并入本文。
第四步:向子嵌段设计变体添加适配子序列-从每个设计中检索所有子嵌段的变体产生序列后添加适配子序列。适配子序列附接至3'和5'末端,以便于从传播载体中释放分配单元,并允许将组合的单元整合到目标载体中。适配子序列定义如下:
5'和3'区段适配子附接至所有区段。所述适配子含有与目的载体整合位点同源的短区(35-250bp)和限制酶识别位点(理想地为iis型限制酶),以允许从克隆载体中释放装配的区段。
5'和3'嵌段适配器附接至所有嵌段。所述适配子含有与目的载体整合位点同源的短区(15-200bp)和限制酶识别位点(理想地为iis型限制酶),以允许从克隆载体中释放装配的区段。
5'和3'区段子嵌段附接至所有子嵌段。所述适配子含有与目的载体整合位点同源的短区(15-100bp)和限制酶识别位点(理想地为iis型限制酶),以允许从克隆载体中释放装配的区段。
根据以下设计规则将适配子序列附接至子嵌段。如果子嵌段的5'序列对应于区段的5'序列,则将5'区段适配子附接至所述子嵌段的5'。如果子嵌段的3'序列对应于区段的3'序列,则将3'区段适配子附接至所述子嵌段的3'。另外,如果子嵌段的5'序列对应于嵌段的5'序列,则将5'嵌段适配子附接至所述子嵌段的5'。如果子嵌段的3'序列对应于嵌段的3'序列,则将3'嵌段适配子附接至所述子嵌段的3'。此外,对于每个子嵌段5'和3',子嵌段适配子被附接至5'和3末端。当附接多个适配子序列时,子嵌段适配子将是最外面的适配子,随后是嵌段适配子,并且在适用时,随后是区段适配子。
在某些实施例中,将包含唯一条形码序列的另外的末端条形码适配子序列添加到子嵌段的两个末端。所述适配子序列含有特异性引物退火位点,用于随后对子嵌段(其充当装配单元以装配单个嵌段)组的并行pcr扩增。给定区段的所有子嵌段在一个末端(5'末端)含有相同的区段特异性条形码序列,而在另一个末端(3'末端)它们含有嵌段特异性条形码序列,以便于从子嵌段文库(从头dna合成后提供)扩增给定嵌段的所有子嵌段。
在某些实施例中,如果线性dsdna子嵌段用作构建嵌段,则可以省略适配子序列。
第五步:同义序列变体池的从头dna合成-通过从头dna合成来合成所有dna子嵌段变体,产生双链dna的文库,每个子嵌段存在于一个或多个同义序列变体中。
由于从头dna合成产率的限制(对于1kb基因合成,大约80%)不是每个子嵌段变体都能成功产生,然而,由于重编码,阻碍特定子嵌段变体的从头dna合成的已知或隐藏序列约束不会跨序列变体传播。增加尝试合成的序列变体的数量将增加可以制造至少一个同义序列变体的可能性。
第六步:连续pcr以分离构建每个嵌段所必需的子嵌段变体组-子嵌段双链dna变体的文库用作模板用于单个子嵌段池的平行pcr扩增。每个pcr扩增的子嵌段池将含有构建特定嵌段所需的所有成功合成的子嵌段序列变体。用于所述子嵌段池的pcr扩增的方法包括用于本领域已知的dna序列扩增的当前pcr方案并且使用
-第一引物,其能够特异性地退火至存在于子嵌段变体的末端区的所述区段特异性条形码序列
-第二引物,其能够退火至存在于子嵌段变体的末端区的所述嵌段特异性条形码序列。
技术人员理解,扩增通过它们的序列必须是可辨别的,即必须选择以允许这种区分的方式放置的pcr引物。
第七步:去除末端pcr条形码序列和构建体的高级装配-在pcr扩增后,通过限制性内切核酸酶消化(bbsl或识别5'和3'子嵌段适配子序列的相似限制酶)释放附接至子嵌段的单个池的末端条形码序列。同义子嵌段的整体同时(在汇集的反应中)使用本领域和使用中已知的同源末端连接装配成随后的高级装配。然后通过限制酶消化(bspqi或识别5'和3'嵌段适配子序列的相似限制酶)从克隆载体释放由此产生的嵌段阵列,并且进一步装配成区段。然后通过限制酶消化(pad、pmel或ceul、seel或识别5'和3'区段适配子序列的相似限制酶)从克隆载体释放产生的区段阵列,并且随后装配成最终的较大(基因组)构建体。由于高级装配不再依赖于每个dna子嵌段变体的成功合成,因此可以快速完成大规模dna设计,允许广泛的遗传零件文库和编码合成途径或整个合成基因组的数千碱基长合成dna构建体的变体的高性价比且高度平行化的装配。
本文描述的方法不依赖于待制造的dna单元的从头dna合成可行性的先前知识。
在某些实施例中,进行非序列验证的合成dna单元以及由成百上千个遗传元件构成的组合零件文库的装配。
无论何处将单个可分离特征的替代物在本文中作为“实施例”列出,应理解此类替代物可以自由组合以形成本文披露的本发明的离散实施例。
实例
概念验证研究的描述:
使用与高通量测序(tnseq)偶联的超饱和转座子诱变,诸位发明人最近鉴定了细胞周期模型生物新月柄杆菌(caulobactercrescentus)的整套必需序列。从这些序列中,诸位发明人已经产生了编码细菌细胞最基本功能的全面的全基因组dna序列(dna零件)列表。特别地,已经为细胞周期模型生物新月柄杆菌定义了覆盖所有必需和高适合度功能的零件列表。多重dna零件定义方法,包括wetlab程序,生物信息学路线和dna序列的重构,描述于(christen,m.,等人http://doi.org/10.1021/acssynbio.5b00087)。该零件列表包含编码必需蛋白质、rna和调节特征的596个单一和复合dna零件。根据柄杆菌属(caulobacter)na1000基因组注释(ncbi登录号:nc_011916.1)加上另外的5'调节序列(启动子)和终止子区,将蛋白质编码基因的零件边界设定为编码序列坐标。根据之前鉴定的必需启动子区设定调节性上游序列的边界(christen,b.,abeliuk,e,,collier,j.m.,kalogeraki,v.s.,passarelli,b.,coller,j,a.,等人(2011).theessentialgenomeofabacterium[细菌的基本基因组].mol.syst.biol.[分子系统生物学],7(1),528-528.http://doi.org/10.1038/msb.2011.58)并且在必要时如由rnaseq确定的扩大到包括强转录起始位点(bozhou,b.,schrader,j.kalogeraki,v.s.,abeliuk,e.,dinh,c.d,,等人(2015).theglobalregulatoryarchitectureoftranscriptionduringthecaulobactercellcycle[在柄杆菌属细胞周期期间转录的总体调节体系结构].,11(1),e1004831.http://doi.org/10.1371/journal.pgen.1004831)。对于必需或高适合度基因,包括预测的rho非依赖性终止子序列(gardner,p.p.,barquist,l,bateman,a.,nawrocki,e.p.,&weinberg,z.(2011).rnie:genome-widepredictionofbacterialintrinsicterminators[rnie:细菌固有终止子的全基因组预测].nucleicacidsresearch[核酸研究],39(14),5845-5852.http://dos.org/10.1093/nar/gkr168)。必需和高适合度dna零件按照在野生型基因组上发现的顺序和方向连接,并编译成773'851碱基对长驯化基因组设计(图2),基因组设计实现强序列重构、部分重构和所有编码序列的完全重编码。序列设计和序列重编码的方法描述于ep15195390.8中,其通过引用以其整体并入本文。
为了定位对于从头dna合成最有问题的序列,将基因组设计分配为三十七个20kb长的基因组区段,其进一步分配为从从头dna合成的商业提供者订购的236个dna构建嵌段(gen9公司剑桥,马萨诸塞州,美国)。从这些中,181个嵌段由gen9公司制造(成功率为75.3%),而55个嵌段从头dna合成失败(表1)。该结果表明,使用低成本的dna从头合成方法,从头dna合成的现有技术状态不能以100%的产率产生每个dna装配单元。
这些序列中被证明最难合成的是区段25(大小为21.3kb),其中6个装配嵌段中的3个从头dna合成失败(表1)。
诸位发明人使用上面概述的多重进化指导的基因组装配策略来执行所述区段25的中性重编码并产生一组3种设计变体。平均而言,每个设计变体含有2'832个碱基取代,对应于13.6%的序列被在开放阅读框(表2)中随机分布的同义密码子取代替换,不包括thr的不可突变区和重叠编码序列。
通过从头dna合成以三种变体制造区段25,以产生作为双链dna的子嵌段变体的文库。在从头dna合成的商业提供者(gen9公司)订购的60个子嵌段中,成功合成了52个,而对于8个子嵌段合成失败(表3和图3a),结果,对于任何单一dna设计没有获得完整的子嵌段组,说明了用于可靠制造双链dna序列的从头dna合成方法的当前缺点。
在5个pcr反应中扩增区段25的所有5个嵌段的子嵌段变体池(图3b)。每个pcr含有一对特异性pcr引物(表6),用于扩增给定嵌段装配所必需的子嵌段的子池。用iis型限制酶(bbsl)消化pcr扩增的子嵌段池以切割pcr适配子序列。每个消化反应包含要装配成给定嵌段的所有四个子嵌段的池,每个子嵌段以三种设计变体表示。这导致区段25的总共五次独立消化反应。将所得线性子嵌段dna的文库装配成它们相应的嵌段,并使用20μl体积的等温装配反应整合到目的载体(pxmcs-2)中。作为对照反应,诸位发明人仅使用来自设计变体1、2或3的子嵌段作为模板对区段25的嵌段#3进行装配反应。没有单独的(不完整的)装配反应产生阳性克隆成功装配嵌段#3。含有嵌段#3的所有子嵌段变体的pcr池产生正确装配的嵌段的阵列,每个嵌段含有子嵌段变体的同义组合(表4),随后将4kbdna嵌段装配成20kb区段并使用酵母重组工程克隆到低拷贝质粒pmr10y中(图3e,3f)。使用标准桑格(sanger)测序对装配的20kb合成区段进行序列验证。
与使用单独添加的子嵌段#1-4的等摩尔比率的对照反应相比,用pcr扩增的子嵌段变体的子池的装配反应产生相当数量的集落(表4)。因为在单个工艺步骤中对于给定的嵌段装配反应,本发明的连续pcr程序扩增含有所有存在的同义子嵌段设计变体的子池,所以不需要从头dna合成子嵌段产率的精细预分析以及广泛的液体处理步骤。
利用适当用于制造大规模dna序列的冗余dna合成策略,以高性价比的方式设计和制造人工生物系统将变得可行。一方面,对于功能性合成基因组设计的完成速度,这将具有深远影响。此外,更大的序列灵活性可实现更显著的序列重构,包括从头dna合成的序列优化、密码子使用适应、遗传密码编辑和cds的重编码,以消除导致dna零件和/或宿主细胞之间干扰的重叠基因调节性特征。此外,用于将相关遗传功能分组在一起以促进共调节和交换的去片段化变得可行(例如将参与脂质代谢、基因组复制和稳定性等的trna或基因分组在一起)。
材料与方法:
合成必需基因组构建体的设计。
使用之前鉴定的必需基因组数据组产生编码新月柄杆菌富培养基生长所需的必需和高适合度功能的dna序列(dna零件)的综合列表(christen,b.,等人http://doi.org/10.1038/msb.2011.58)dna零件列表包括编码蛋白质、rna和调节性特征的dna序列以及小的必需的基因间序列。根据新月柄杆菌na1000基因组注释(ncbi登录号:nc_011916.1)加上另外的5'调节序列(启动子)和终止子区,将蛋白质编码基因的零件边界设定为cds坐标。根据之前鉴定的必需启动子区设定必需基因的调节性上游序列的边界,并且在必要时如由rnaseq确定的扩大到包括强转录起始位点。对于必需或高适合度基因,包括预测的rho非依赖性终止子序列。基本和高适合度dna零件按照在野生型基因组上发现的顺序和方向连接,并编译成773'354碱基对长合成基因组构建体。然后将该基因组构建体分配为38个20kb长的区段(图3)
驯化基因组设计的序列优化和变体产生。
为了优化合成基因组区段的序列,通过中性重编码(同义密码子替换)重构蛋白质编码序列,以消除已知抑制大规模从头dna合成的不允许的序列模式。跨区段的平均重编码概率设定为0.57,导致跨越773851bp基因组设计引入133354个碱基取代。cds的前四个氨基酸密码子被排除在重编码之外以维持潜在的翻译和其他调节信号。在重编码后去除的不允许的序列包括bsal、aarl、bbsl、bspqi、pad和pmel、seel和ceul的内切核酸酶位点。此外,agt、ata、aga、gta和agg密码子(它们是新月柄杆菌中的稀有密码子)被设定为不可突变密码子(在重编码后既不替换也不引入)。在重编码后,琥珀终止密码子tag和亮氨酸的两个tta和ttg密码子被消除。去除均聚序列和二核苷酸和三核苷酸重复的发生(少于6个g、8个c'、9个a或t、少于10个重复的二核苷酸、少于6个重复的三核苷酸)。相似地,去除了大于11bp的直接和间接序列重复。为了产生片段25的变体设计,执行天然序列设计的第一次重编码以去除任何合成约束。gc和at含量在99bp窗口内设定为不超过70%,并且在21bp窗口内不超过85%。为了产生区段25的随后设计变体,将总体重编码概率设定为0.4。对于设计变体1,对于99bp和21bp窗口大小gc和at限制分别设定为0.62和0.8,对于设计变体2,对于99bp和21bp窗口大小gc和at限制设定为0.58和0.75,并且对于设计变体3,对于99bp和21bp窗口大小gc和at限制分别设定为0.54和0.70。
子嵌段池的平行pcr扩增
涵盖区段25的设计变体的子嵌段序列包含在pg9m-2低拷贝数质粒文库中,该质粒文库代表已经成功制造的子嵌段形式区段25的所有设计变体(表3和图3)。使用
子嵌段和pxmcs-2靶标载体的消化
用bbsl型iis限制酶消化pcr扩增的子嵌段池。每个消化反应含有相应嵌段的所有四个子嵌段变体的池,导致对于区段25总共五个独立的消化反应。随后在20μl反应体积中对五个子嵌段池中的每一个进行消化,该反应体积含有:10μl的直接取自pcr反应混合物的子嵌段池、0.5μl(5u)bbsl型iis限制酶(neb,美国)、2μl10xnebuffer2.1(neb,美国)和7.5μl无核酸酶h20(普洛麦格公司(promega),usa)。将消化反应在37℃孵育过夜,并且随后经柱纯化,并且使用
在40μl消化反应体积中用ndel和nhel-hf限制酶消化pxmcs-2靶标载体,该消化反应体积由以下构成:20μl(294,4ng/μl)pxmcs-2、0.5μl(10u)ndel(neb,美国)、0.5μl(10u)nhel-hf(neb,美国)、4μl10x
子嵌段的dna装配成嵌段:
将bbsl消化的子嵌段池装配成它们相应的嵌段并在等温20μl装配反应中整合到它们的靶标载体pxmcs-2中,该反应使用:4μl5x等温反应缓冲液、0.008μl(0.08u)t5外切核酸酶(neb,美国)、0.25(2.5u)
将装配的嵌段电穿孔到大肠杆菌(e.coli)中
取出5μl的每种pxmcs-2::嵌段[0-4]装配体,并在0.025μm的vswpmftm膜过滤器(默克密理博有限公司(merckmilliporeltd.),irl出版社)上透析20min。随后,使用0.1cm电极间隙基因
对子嵌段连接进行pcr以验证嵌段装配
使用基因组分配器的自动设计的引物组验证正确的嵌段装配(表7)。通过菌落pcr直接从含有pxmcs-2::嵌段[0-4]的大肠杆菌dh5a扩增子嵌段连接。挑取菌落并在补充有卡那霉素(20μg/ml)的液体lb肉汤中生长。使用液体培养物作为模板进行每个嵌段的子嵌段连接的pcr扩增。在20μl最终反应体积中,添加10μl2x
bspqi介导的从pxmcs-2载体释放嵌段
使用genejet质粒微量制备(miniprep)试剂盒(赛默飞世尔科技公司,美国)从各自的dh5a菌株(参见菌株,bc3744-bc3748,表8)中纯化质粒pxmcs-2::嵌段[0-4]。随后,经由bspqi型iis限制性消化从pxmcs-2骨架释放嵌段(图3c)。每个嵌段释放由40μl消化反应体积组成,该反应体积由以下构成:10μl(>5μg)pxmcs-2::嵌段[0-4]质粒、1μl(10u)bspqi型ns限制酶(neb,美国)、4μl10xnebuffer3.1(neb,美国)和25μl无核酸酶h2o(洛麦格公司,美国)。该消化在50℃孵育1.5h,并且随后经由在80℃孵育20min停止反应。使用
来自嵌段[0-4]的区段的酵母装配
柱纯化的嵌段[0-4]用于将区段25装配进入pmr10y(pmr10::cen/ars::ura3)质粒骨架。使酿酒酵母(s.cerevisiae)菌株vl6-48n(bc3347)生长至od6000.7,其中沉淀2ml,并且然后重悬于1ml0.9%nacl溶液中。再次沉淀培养物,弃去nacl溶液上清液并添加100μg鱼精子dna(来自鲑鱼睾丸的单链,d7656,西格玛奥德里奇公司(sigma-aldrich),美国)。随后,将-540μg线性化的pmr10y和300μg的每种嵌段消化物添加到沉淀中。彻底涡旋后,将沉淀重悬于500μl转化混合物(400μl50%peg溶液、50μl1m乙酸锂、50μlddh2o)中。为了完成转化,将57μldmso添加到转化反应中并且在室温下孵育15min,随后直接在42°下热激孵育15min。最后,将培养物沉淀,弃去上清液,将沉淀重悬于100μlddh2o中并接种在酵母合成营养缺陷型培养基上(不含尿嘧啶,+葡萄糖(10g/l),+腺嘌呤(80mg/l)并在30℃下孵育三天。
酵母菌落pcr以验证区段25嵌段连接
使用基因组分配器的自动设计的引物组,通过对来自上述装配步骤的转化的酵母菌落pcr直接扩增每个嵌段连接以验证正确的区段装配。挑取六个菌落并在液体酵母合成营养缺陷型培养基(不含尿嘧啶,+葡萄糖(10g/l),+腺嘌呤(80mg/l)中生长。用于扩增嵌段连接的pcr在20μl如下反应体积中进行:10μl2xphire绿色热起始iipcr反应混合物(greenhotstartiipcrmastermix)(赛默飞世尔科技公司,美国)、0.5μl25μm正向引物(#33-40的fw引物)、0.5μl25μm反向引物(#33-40的rv引物)、1转化的酵母液体培养物和8μlddh2o。该pcr方案由以下组成:(1)在98℃下初始变性3:00min,(2)在98℃下变性5s,(3)在62℃下引物退火5s,(4)在72℃下延伸20smin,(5)重复步骤2-4,40次,(6)在72℃下最终延伸1min
使用的分配参数、dna适配子序列和条形码。
应用了以下分配参数:区段大小:20'000bp,区段重叠:120bp,嵌段大小:4'000bp,嵌段重叠:80bp,子嵌段大小1'000bp,子嵌段重叠:25bb。用于分配的适配子序列列于表5中,用于亚池pcr扩增的条形码引物列于表6中。用于嵌段装配的pcr验证的引物列于表7中。
表1:被分配为4kb嵌段的驯化基因组的从头dna合成产率
表1:表头具有以下含义:区段:如在驯化基因组设计中注释的区段编号,坐标:根据基因组设计的genbank文件的碱基对序列坐标,大小[bp]:以碱基对表示的区段的长度,嵌段:每个区段使用的分配嵌段的数目,合成失败:在第一轮从头dna合成期间合成失败的嵌段列表,产率[%]:从头dna合成成功的区段序列的百分比。
表2:区段25的子嵌段变异体设计之间的碱基取代率
表2:表头具有以下含义:sbid:如在驯化基因组设计中注释的子嵌段编号,坐标:根据基因组设计的genbank文件的碱基对序列坐标,大小[bp]:以碱基对表示的区段的长度,碱基取代率:在设计变体之间发生的子嵌段的碱基取代数,开始:子嵌段起始位置的基因组坐标,结束:子嵌段终点位置的基因组坐标,大小[bp]:以碱基对表示的子嵌段的大小。
表3:来自区段25的3个子嵌段设计变体的从头dna合成产率
表3:对于在3个同义设计变体中构建区段25的60个子嵌段中的8个从头dna合成失败。所有设计变体都没有产生成功装配区段25所需的所有子嵌段。表头具有以下含义:设计:序列设计变体,嵌段:嵌段编号,子嵌段:子嵌段编号,长度:通过从头dna合成产生的子嵌段大小,产率(ng):以纳克的dna表示的质粒克隆的子嵌段的产率,菌株id:菌株标识号。
表4:使用子嵌段变体池的嵌段装配反应的效率
表4:表头具有以下含义:装配反应:装配反应的名称,子嵌段设计变体:在装配反应期间使用的特定子嵌段的一个多个设计变体。sb:子嵌段编号,菌落数:电穿孔后获得的菌落和相应的dh5apxmcs-2::嵌段[0-4]装配体的结果,a具有仅消化的子嵌段和消化的pxmcs-2进入大肠杆菌dh5a的对照反应分别产生0和8个菌落,b通过pcr确认嵌段1的3个克隆中的2个。
表5:用于分配的适配子序列的列表
表5:适配子:适配子的类型,序列:适配子dna序列。
表6:子池pcr扩增使用的条形码引物的列表
表6:条形码#:条形码id,引物:引物的名称,序列:寡核苷酸引物的dna序列。
表7:用于嵌段装配的pcr验证的引物的列表
表7:引物#:引物编号,引物id:引物的名称,连接:子嵌段连接的名称,序列:引物dna序列。
表8:菌株列表:
表头具有以下含义:菌株:菌株的名称,描述:菌株和基因型的描述,*larionov,v.,kouprina,n.,nikolaishvili,n.,&resnick,m.a.(1994).recombinationduringtransformationasasourceofchimericmammalianartificialchromosomesinyeast(yacs)[转化期间的重组作为酵母中嵌合哺乳动物人工染色体(yac)的来源].nucleicacidsresearch[核酸研究],22(20),4154-4162。
- 还没有人留言评论。精彩留言会获得点赞!