一种贝他斯汀药物中间体的制备工艺的制作方法
本发明属于有机合成技术领域,具体涉及一种贝他斯汀中间体的制备工艺。
背景技术:
苯磺酸贝他斯汀(bepotastinebesilate),化学名为4-[(s)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶丁酸单苯磺酸盐,结构如式(i)所示。它是由日本tanabeseiyaku公司和ubeindustries公司联合开发并于2000年7月和2002年1月先后被批准用于治疗过敏性鼻炎、荨麻疹、皮肤病引起的瘙痒症和神经性耳鸣,商品名为talion。2009年8月,ista制药公司获fda批准生产苯磺酸贝他斯汀滴眼液,商品名bepreve,用于治疗过敏性结膜炎相关性眼瘙痒(exp.eyeres.2010,91,85;foliapharmacol.jpn.1997,110,19;j.dermatol.sci.2003,32,237.)。研究发现s构型的苯磺酸贝斯他汀要比r构型的对映异构体具有更高的药效活性,且毒副作用更小(jp10-237070或jp1998237970)。本品对组胺h1受体具有选择性的结合抑制作用,同时具有稳定肥大细胞功能的能力,作用迅速且选择性强,无其他抗过敏药的镇静不良反应,疗效好,临床应用前景广阔(clinophthalmol,2014,8,1495-1505.)。
原研公司tanabeseiyaku和ubeindustries在专利jp1998237070、jp2000198784和wo9829409公布了通过外消旋拆分法制备(s)-贝他斯汀合成路线。苯磺酸贝他斯汀的工艺合成关键在于构建(s)-(4-氯苯基)(吡啶-2-基)甲醇(iii),而手性拆分是目前工业化生产的主流方法(wo2008153289)。拆分副产物r构型异构体成为废弃品,导致整条路线生产成本增高,三废排放量增多。鲁南制药集团有限公司报道了利用不对称催化氢化合成贝他斯汀的路线(中国医药工业杂志,2006,37,726-727),他们以(s)-[ru(binap)cl2]2(net3)作为手性催化剂,经(4-氯苯基)(吡啶-2-基)甲酮(ii)的不对称氢化得到(s)-(4-氯苯基)(吡啶-2-基)甲醇(iii),ee值达98.5%。虽然此路线避免了手性拆分这一工序,但是由于催化剂效率较差,ton仅为186,且催化剂对空气敏感,严重限制了其在工业制药上的应用。宜昌人福药业有限责任公司开发了一条不对称氢转移法合成贝他斯汀的方法(cn106938995a)。该法利用(4-氯苯基)(吡啶-2-基)甲酮氮氧化物为起始原料,经不对称氢转移法还原成(s)-(4-氯苯基)(吡啶-2-基)甲醇-n-氧化物,最后再经过一次还原制得贝他斯汀中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇,产物收率达到95%,ee值为92%。不对称氢转移过程中使用的催化剂用量较大,ton仅为20,路线十分繁琐,生产成本大幅度提高,使得难以在工业规模上应用。目前国内还没有厂家规模化生产苯磺酸贝他斯汀原料药产品,主要依靠进口。因此,开发一条具有工业化应用前景的苯磺酸贝他斯汀关键中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇的不对称合成新工艺具有很高的社会效益和经济效益。
技术实现要素:
针对现有技术存在的上述技术问题,本发明的目的在于提供一种贝他斯汀药物中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇的制备工艺。
所述的一种如式(iii)所示的贝他斯汀药物中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇的制备工艺,其特征在于包括如下步骤:
1)在氩气氛围及0℃~60℃的反应温度下,将金属m络合物与手性配体l*依次加入到溶剂a中,搅拌反应0.5~6小时,制得金属m与手性配体l*配位结合的催化剂[m]/l*,所述金属m为ru、rh、ir或pd;
2)向高压釜中依次加入式(ii)所示的(4-氯苯基)(吡啶-2-基)甲酮、步骤1)所得催化剂[m]/l*、溶剂b及碱,于0℃~100℃温度以及0.1~10.0mpa的氢气压力下进行不对称加氢反应,反应时间为2~24小时,反应结束后,反应液减压浓缩回收溶剂b,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得式(iii)所示的(s)-(4-氯苯基)(吡啶-2-基)甲醇;
具体反应路线如下:
所述的手性配体l*的化学结构式如通式(1)或通式(2)所示:
通式(1)中:r1和r2各自独立的选自c1~c6的烷基、c3~c6的环烷基、芳基或杂环芳基;r3为芳基、杂环芳基或c1~c6的烷基;y为(ch2)n,其中n为1~6的整数;
通式(2)中:r1和r2各自独立的选自c1~c6的烷基、c3~c6的环烷基、芳基或杂环芳基;r3为芳基、杂环芳基或c1~c6的烷基;r4为c1~c6的烷基、芳基、杂芳基或氢。
所述的一种贝他斯汀药物中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇的制备工艺,其特征在于步骤1)中,所述手性配体l*的化学结构式为如式l1~l20中的任意一种所示;
所述的一种贝他斯汀药物中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇的制备工艺,其特征在于所述金属m络合物为pd(cod)cl2、pd(pph3)4、pdcl2(pph3)2、pd(dba)2、pd(oac)2、pdcl2l2、[rh(nbd)2]+bf4、[rh(nbd)cl]2、[rh(cod)cl]2、[rh(cod)2]x、[rh(acac)(co)]2、rh(ethylene)2(acac)、rh(ethylene)2cl2、rhcl(pph3)3、rh(co)2cl2、ruhx(l)2(diphosphine)、ru(arene)x2(diphosphine)、ru(ar)x2、ru(rco2)2(diphosphine)、ru(methallyl)2(diphosphine)、ru(arylgroup)x2(pph3)3、rux2(l)2(diphosphine)、ru(cod)(cot)、ru(cod)(cot)x、rux2(cymene)、ru(arylgroup)x2(diphosphine)、rucl2(cod)、[ru(cod)2]x、rux2(diphosphine)、ru(arh)cl2、ru(cod)(methallyl)2、[ir(nbd)2cl]2、ir(nbd)2)x、[ir(cod)cl]2或[ir(cod)2]x中的任意一种,其中r为c1~c6的烷基或烷氧基;其中aryl为芳基,x为阴离子,x优选为bf4-、clo4-、sbf6-、pf6-、cf3so3-或b(ar)4-,ar为二(三氟甲基)苯或氟苯,l选自乙腈或苯甲腈。
所述的一种贝他斯汀药物中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇的制备工艺,其特征在于步骤1)中的溶剂a和步骤2)中的溶剂b各自独立的选自二氯甲烷、四氢呋喃、2-甲基四氢呋喃、甲基叔丁基醚、甲苯、甲醇、乙醇、正丙醇、异丙醇、叔丁醇中的一种或两种以上混合溶剂,溶剂a和溶剂b相同或不同。
所述的一种贝他斯汀药物中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇的制备工艺,其特征在于步骤1)中,反应温度为10℃~40℃。
所述的一种贝他斯汀药物中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇的制备工艺,其特征在于步骤2)中,催化剂[m]/l*、碱与(4-氯苯基)(吡啶-2-基)甲酮的摩尔比为1:10~200:100~100000。
所述的一种贝他斯汀药物中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇的制备工艺,其特征在于步骤2)中,进行不对称氢化反应温度为10℃~60℃。
所述的一种贝他斯汀药物中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇的制备工艺,其特征在于步骤2)中,进行不对称氢化反应的氢气压力为1.0~5.0mpa。
所述的一种贝他斯汀药物中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇的制备工艺,其特征在于步骤2)进行不对称氢化反应中,(4-氯苯基)(吡啶-2-基)甲酮在溶剂b中的浓度为0.05mol/l~5.0mol/l,优选为0.1~1.0mol/l。
所述的一种贝他斯汀药物中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇的制备工艺,其特征在于步骤2)中,所述碱为叔丁醇钾、叔丁醇钠、叔丁醇锂、碳酸铯、碳酸钾、碳酸钠、甲醇钠、氢氧化钠或氢氧化钾。
通过采用上述技术,与现有技术相比,本发明具有以下优点:
1)本发明所采用的手性二茂铁配体合成简单、价格便宜,对空气和水不敏感,可以稳定存在。本发明的方法反应条件温和,操作简便,易于工业化生产,具有较大的实施价值和社会经济效益;本发明制备金属m与手性配体l*配位结合的催化剂[m]/l*的过程中,制得的催化剂[m]/l*不需进行特别的提纯处理,可直接用于催化加氢制备如式(iii)所示的贝他斯汀药物中间体(s)-(4-氯苯基)(吡啶-2-基)甲醇的反应;
2)本发明所用的手性配体与ir、rh、pd等金属形成的催化剂具有较高的活性和对映选择性,能将(4-氯苯基)(吡啶-2-基)甲酮还原为(s)-(4-氯苯基)(吡啶-2-基)甲醇,定量转化,且ee值高达99.9%。此外,与传统的手性拆分法制备(s)-(4-氯苯基)(吡啶-2-基)甲醇的工艺相比,该方法具有催化剂用量低,收率高等特点,该催化剂用量可以降至万分之一以下,能公斤级地制备(s)-(4-氯苯基)(吡啶-2-基)甲醇。
附图说明
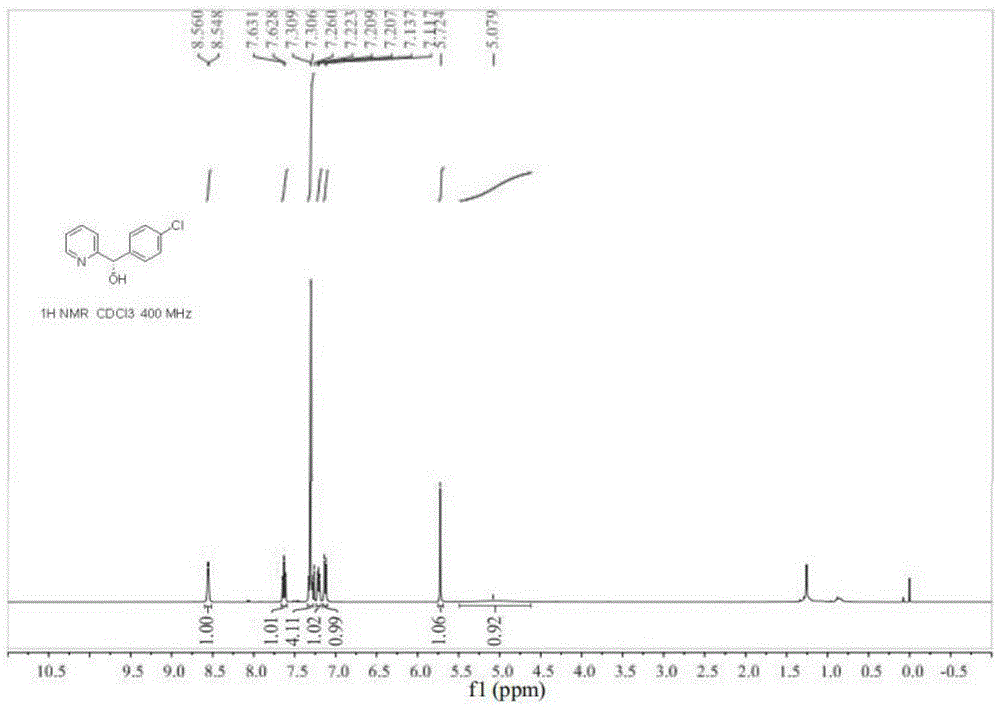
图1为实施例12制备的(s)-(4-氯苯基)(吡啶-2-基)甲醇产品的1hnmr谱图;
图2为实施例12制备的(s)-(4-氯苯基)(吡啶-2-基)甲醇产品的13cnmr谱图;
图3为消旋体(4-氯苯基)(吡啶-2-基)甲醇的hplc分析谱图;
图4为实施例12制备的(s)-(4-氯苯基)(吡啶-2-基)甲醇产品的hplc分析谱图。
具体实施方式
下面结合具体实施例对本发明作进一步说明,但本发明的保护范围并不限于此。
实施例1:
(1)将手性配体l1(16.6mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂、叔丁醇钾(1.34g,12mmol)和甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(51.0g,0.23mol),收率:97%,hplc纯度为98%,ee值为96%。
实施例2:
(1)将手性配体l1(16.6mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂、叔丁醇锂(0.96g,12mmol)和甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(51.2g,0.23mol),收率:97%,hplc纯度为97%,ee值为99%。
实施例3:
(1)将手性配体l1(16.6mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,碳酸钠(1.3g,12mmol),甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(46.1g,0.21mol),收率:88%,hplc纯度为98%,ee值为91%。
实施例4:
(1)将手性配体l1(16.6mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,甲醇钠(0.65g,12mmol),甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(49.0g,0.22mol),收率:93%,hplc纯度为98%,ee值为89%。
实施例5:
(1)将手性配体l1(16.6mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入异丙醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(51.1g,0.23mol),收率:97%,hplc纯度为97%,ee值为98%。
实施例6:
(1)将手性配体l1(16.6mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),甲醇(100ml),充入h2(5.0mpa),40℃下反应8h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(52.0g,0.24mol),收率:99%,hplc纯度为97%,ee值为98%。
实施例7:
(1)将手性配体l1(16.6mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),异丙醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(51.1g,0.23mol),收率:97%,hplc纯度为97%,ee值为99%。
实施例8:
(1)将手性配体l1(16.6mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),甲醇(100ml),充入h2(3.0mpa),60℃下反应8h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(52.0g,0.24mol),收率:99%,hplc纯度为97%,ee值为95%。
实施例9:
(1)将手性配体l1(16.6mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),甲醇(100ml),充入h2(3.0mpa),80℃下反应8h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(52.0g,0.24mol),收率:99%,hplc纯度为97%,ee值为93%。
实施例10:
(1)将手性配体l1(16.6mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),10℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂、叔丁醇钾(1.34g,12mmol)和甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(40.6g,0.19mol),收率:77%,hplc纯度为98%,ee值为94%。
实施例11:
(1)将手性配体l1(16.6mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),40℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂、叔丁醇钾(1.34g,12mmol)和甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(45.8g,0.21mol),收率:87%,hplc纯度为97%,ee值为95%。
实施例12:
(1)将手性配体l2(17.3mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0g,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(50.5g,0.23mol),收率:96%,hplc纯度为98%,ee值为99.9%。
本实施例制备的(s)-(4-氯苯基)(吡啶-2-基)甲醇的1hnmr谱图和13cnmr谱图分别如图1和图2所示,从图1和图2可以确定得到的(s)-(4-氯苯基)(吡啶-2-基)甲醇产品。
消旋体化合物(4-氯苯基)(吡啶-2-基)甲醇和实施例10制备的(s)-(4-氯苯基)(吡啶-2-基)甲醇产品的hplc分析谱图分别如图3和图4所示,对比图3和图4可以看出,(4-氯苯基)(吡啶-2-基)甲醇的两种消旋体在hplc分析谱图中的出峰时间存在差异,可以确定实施例10最终制得的是(s)-(4-氯苯基)(吡啶-2-基)甲醇,产品纯度很高。
实施例13:
(1)将手性配体l4(14.7mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(48.3g,0.22mol),收率:92%,hplc纯度为98%,ee值为93%。
实施例14:
(1)将手性配体l7(17.3mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(48.0g,0.23mol),收率:91%,hplc纯度为98%,ee值为98%。
实施例15:
(1)将手性配体l12(16.9g,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(46.0g,0.20mol),收率:87%,hplc纯度为98%,ee值为67%。
实施例16:
(1)将手性配体l13(16.1g,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(44.2g,0.20mol),收率:84%,hplc纯度为98%,ee值为90%。
实施例17:
(1)将手性配体l15(19.0g,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(48.4g,0.22mol),收率:92%,hplc纯度为98%,ee值为89%。
实施例18:
(1)将手性配体l20(13.4mg,0.025mmol),金属络合物[ir(cod)cl]2(8.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(46.2g,0.21mol),收率:88%,纯度为96%,ee值为71%。
实施例19:
(1)将手性配体l2(0.35g,0.50mmol),金属络合物[ir(cod)cl]2(0.16g,0.24mmol),加入反应瓶中,氩气氛围下加入甲醇(20ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(1.04kg,4.80mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(19.2g,240mmol),甲醇(2.0l),充入h2(5.0mpa),40℃下反应24h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(1.02kg,4.66mol),收率:97%,hplc纯度为97%,ee值为99%。
实施例20:
(1)将手性配体l2(0.035g,0.050mmol),金属络合物[ir(cod)cl]2(0.016g,0.024mmol),加入反应瓶中,氩气氛围下加入甲醇(20ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(1.04kg,4.80mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(19.2g,240mmol),甲醇(2.0l),充入h2(8.0mpa),60℃下反应36h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(1.02kg,4.66mol),收率:97%,hplc纯度为97%,ee值为98%。
实施例21:
(1)将手性配体l2(17.3mg,0.025mmol),金属络合物rh(cod)2bf4(5.0mg,0.012mmol),加入反应瓶中,氩气氛围下加入甲醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(47.3g,0.21mol),收率:89%,hplc纯度为97%,ee值为92%。
实施例22:
(1)将手性配体l2(17.3mg,0.025mmol),金属络合物pd(oac)2(2.7mg,0.012mmol),加入反应瓶中,氩气氛围下加入异丙醇(1.5ml),25℃下搅拌反应0.5h,制得催化剂。
(2)在高压釜中加入(4-氯苯基)(吡啶-2-基)甲酮(52.2g,0.24mol),直接加入步骤(1)所制备的催化剂,叔丁醇锂(0.96g,12mmol),甲醇(100ml),充入h2(3.0mpa),40℃下反应12h,反应结束后,反应液减压浓缩回收有机溶剂,再加入适量水,用乙酸乙酯萃取,分液为有机相和水相,有机相经干燥、脱溶,制得(s)-(4-氯苯基)(吡啶-2-基)甲醇(35.0g,0.16mol),收率:67%,hplc纯度为95%,ee值为74%。
本说明书所述的内容仅仅是对发明构思实现形式的列举,本发明的保护范围不应当被视为仅限于实施例所陈述的具体形式。
- 还没有人留言评论。精彩留言会获得点赞!