一种多取代异吲哚类化合物及其制备方法与流程
本发明涉及有机合成领域,尤其涉及一种多取代异吲哚类化合物及其制备方法。
背景技术:
异吲哚及其衍生物同其他氮杂环化合物一样,广泛存在于多种具有显著生物活性的天然产物和药物分子之中。异吲哚为核心骨架也是许多生物活性化合物和天然产物共有的结构砌块,合成更多有良好治疗效果的药物也吸引着众多科学家,同时异吲哚类化合物在精细化工领域也有广泛的应用。例如1982年从海绵中分离出第一种天然存在的对微生物具有一定抗菌作用的异吲哚类化合物。随后越来越多的含异叫睬的天然产物被人们所发现。星型抱菌素被发现并归类于异吲哚并咔唑类生物碱,在过去的几十年中超过60种的叫异吲哚并咔唑已经从几种细菌海洋无脊椎动物中分离出。至今为止,星形抱菌素都是最有效的蛋白激酶抑制剂之一,也在临床试验中用于治疗急性骨髓白血病。同时,异吲哚的生物碱和阿扑吗啡生物碱也相继从植物中分离得出,数量庞大,种类繁多。其次异吲哚类化合物在有机颜料中也具有其重要的价值如拜耳公司通过生产方法的改进的异吲哚衍生物制备成色光艳丽和高纯的颜料诸如pigmentyellow139,pigmentorange66,pigmentbrown38等。但是它们在自然界存在的提取量及产率均不能满足人们的需要,因此高效、模块化合成该类异吲哚具有良好价值。
异吲哚骨架的经典合成方法有以下几种:1:以1,2,3,4-四氢化萘-1,4-亚胺为原料通过高温加热、液氮冷却等步骤合成了异吲哚。2:通过1,2一二溴苯为原料通过烷基化反应制得异吲哚琳然后高温加热制得了异吲哚。3:以邻苯二甲醛为原料与氯化按和硫基乙醇反应制备了了异吲哚。这些传统的合成方法在产率规模化生产虽然值得借鉴,但是其步骤复杂,合成的副产物污染性相对严重不利于绿色友好化学的发展趋势。其中季碳中心的异吲哚类化合物常规方法难以制备,季碳上的取代物易转化被格氏试剂类亲核取代等多种转化模式。
而在过去的20年间,通过过渡金属催化的c-h键芳基化反应已经成为构筑联多环化合物最有效的合成策略之一,特别是铑对催化芳基c-h活化的研究较为广泛。它可以有效的缩短合成步骤,减少对环境的污染。另一方面,heck反应作为钯催化的芳基卤衍生物与末端烯烃的碳-碳键偶联反应,鉴于过渡金属催化的碳氢键活化的发展,人们已经发展一些直接使用未经官能团化的烃类的碳氢键作为底物,实现了烃类碳氢键与末端烯烃的氧化heck反应(oxidativeheckreaction)来直接构建烯烃衍生物。尽管氧化heck反应取得了长足的发展,其成键模式和产物类型还需要进一步反应,比如通过串联反应来丰富成键的多样性、以及产物的类型。
技术实现要素:
本发明提供了一种多取代异吲哚类化合物及其制备方法,解决了现有的氧化heck反应的成键多样性和产物类型较低的技术问题。
本发明提供了一种多取代异吲哚类化合物,其结构如式i所示:
本发明还提供了一种多取代异吲哚类化合物的制备方法,将式ii所示化合物与式iii所示化合物溶于惰性溶剂,在催化剂的作用下进行反应,得到式i所示化合物:
其中ar为取代的苯环或杂环类化合物(如取代的噻吩、呋喃、吲哚、吡咯和喹啉),r1为含卤素、酯基、羰基、氨基、硝基、氰基、砜基或酰基的直链烃基、环烃基或稠环芳基;r2、r3和r4为烷基或芳基。
优选的,所述惰性溶剂包括甲苯、四氢呋喃、1,4-二氧六环、n,n’-二甲基甲酰胺、n,n’-二甲基乙酰胺、n-甲基吡咯烷酮、二甲亚砜、乙腈、1,2-二氯乙烷、乙醇和水中的一种或多种。
优选的,所述催化剂为三价铑催化剂。
优选的,所述三价铑催化剂包括五甲基环戊二级氯化铑二聚体、五甲基环戊二烯氯化铱二聚体、三乙腈-五甲基环戊二烯基氯化铑二聚体中的一种或多种。
优选的,还包括添加剂;所述添加剂为六氟锑酸银和/双三氟甲磺酰亚胺银。
优选的,还包括氧化剂,所述氧化剂包括醋酸钠、醋酸银、碳酸银、三氟磺酸银、硝酸银、醋酸铜、卤化亚铜、卤化铜、三卤化铁和硝酸铁钟的一种或多种。
优选的,式ii所示化合物与式iii所示化合物的摩尔比为1:1~1:3。
优选的,所述催化剂与式ii所示化合物的摩尔比为0.5~1。
优选的,所述惰性溶剂的体积为1~2ml,所述反应的温度为80℃~120℃。
本发明涉及一种多取代异吲哚类化合物及其制备方法,其利用三价铑催化剂催化的亚胺酯与烯烃酯类化合物进行碳氢键活化串联反应,实现含有季碳中心异吲哚类化合物类衍生物的高效合成方法。本发明公开了一种在简单的一锅法条件下,通过三价铑催化的亚胺酯与烯烃酯类化合物的氧化heck反应串联反应,得到一种多取代含有季碳中心异吲哚类化合物。而季碳中心的异吲哚类化合物常规方法难以制备,季碳上的取代物易转化被格氏试剂类亲核取代等多种转化模式。该方法具体地就是在惰性溶剂条件下,通过三价铑催化剂与醋酸钠和醋酸铜共同促进下,实现快速构建各种多取代的异吲哚类化合物类衍生物。
本发明实施例提供的制备方法具有高效、良好的化学选择性、区域选择性,同时该方法仅使用简单易得的亚胺烯烃酯类化合物作为反应底物,步骤少、操作简便,而且对底物的适用范围非常地广,也易于后续的进一步转化,该申请发明具有良好的原子经济性。更为重要的是,通过调节底物三组分的结构,该转化可以模块化合成在生物、药物及天然产物领域具有良好的应用价值的多取代异吲哚类化合物。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。
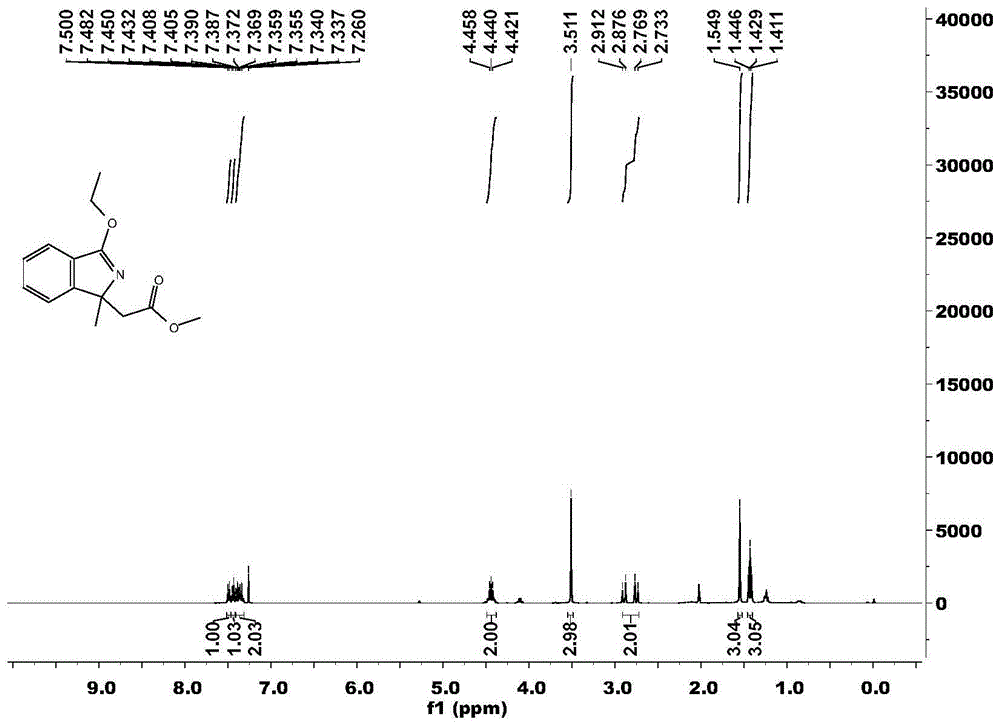
图1为本发明实施例中3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸甲酯的核磁共振氢谱图;
图2为本发明实施例中3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸甲酯的碳-13核磁共振谱;
图3为本发明实施例中3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸苯酯的核磁共振氢谱图;
图4为本发明实施例中3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸苯酯的碳-13核磁共振谱;
图5为本发明实施例中6-溴-3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸甲酯的核磁共振氢谱图;
图6为本发明实施例中6-溴-3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸甲酯的碳-13核磁共振谱;
图7为本发明实施例中6-溴-3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸苯酚酯的核磁共振氢谱图;
图8为本发明实施例中6-溴-3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸苯酚酯的碳-13核磁共振谱;
图9为本发明实施例中3-乙氧基-1-甲基-6-硝基-1h-异吲哚-1-基乙酸甲酯的核磁共振氢谱图;
图10为本发明实施例中3-乙氧基-1-甲基-6-硝基-1h-异吲哚-1-基乙酸甲酯的碳-13核磁共振谱;
图11为本发明实施例中3-乙氧基-6-碘-1-甲基-1h-异吲哚-1-基乙酸甲酯的核磁共振氢谱图;
图12为本发明实施例中3-乙氧基-6-碘-1-甲基-1h-异吲哚-1-基乙酸甲酯的碳-13核磁共振谱;
图13为本发明实施例中5-乙氧基-7-甲基-1-甲苯磺酰-1,7-二氢吡咯[3,4-f]7-吲哚基乙酸甲酯的核磁共振氢谱图;
图14为本发明实施例中5-乙氧基-7-甲基-1-甲苯磺酰-1,7-二氢吡咯[3,4-f]7-吲哚基乙酸甲酯的碳-13核磁共振谱。
具体实施方式
本发明实施例提供了一种多取代异吲哚类化合物及其制备方法,解决了现有的氧化heck反应的成键多样性和产物类型较低的技术问题。
为使得本发明的发明目的、特征、优点能够更加的明显和易懂,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,下面所描述的实施例仅仅是本发明一部分实施例,而非全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
实施例13-乙氧基-1-甲基-1h-异吲哚-1-基乙酸甲酯(1a)
在一个大气压空气氛围下,向15mlschlenk反应管中依次加入苯甲亚胺酸乙酯化合物2a(30.0mg,0.20mmol),2-丁烯酸甲酯3a(40.0mg,0.40mmol),三价铑催化剂三乙腈五甲基环戊二烯氯化铑二聚体[cp*rh(ch3cn)3cl2]2(2.0mg,0.004mmol),三氟甲磺酰亚胺银(6.0mg,0.008mmol),醋酸铜(15.0mg,0.06mmol),醋酸钠(8.0mg,0.06mmol),1,2-二氯乙烷(dce,1ml),在温度为80℃中反应4小时。反应结束后冷却至室温,经硅藻土抽滤后,浓缩得到粗产物。粗产物用制备的硅胶板进行层析色谱分离,所选展开剂或洗脱剂为石油醚与乙酸乙酯的体积比20:1,得到产物3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸甲酯(1a):1hnmr(400mhz,cdcl3)δ7.49(d,j=7.2hz,1h),7.44(d,j=7.2hz,1h),7.41-7.34(m,2h),4.48-4.40(m,2h),3.51(s,3h),2.89(d,j=14.4hz,1h),2.75(d,j=14.4hz,1h),1.55(s,3h),1.43(t,j=6.8hz,3h).13cnmr(100mhz,cdcl3)δ170.7,167.8,156.2,132.1,129.3,127.5,121.5,120.8,69.9,63.9,51.2,43.6,25.3,14.3.
本发明实施例的化学转化提供了一种快速构建多取代异喹啉衍生物,该类分子同时包含乙氧基和酯基具有进一步构建复杂稠环分子的潜能。
实施例23-乙氧基-1-甲基-1h-异吲哚-1-基乙酸苯酯(1b)
在一个大气压空气氛围下,向15mlschlenk反应管中依次加入苯甲亚胺酸乙酯化合物2a(30.0mg,0.20mmol),2-丁烯酸苯酚酯3b(84.0mg,0.50mmol),三价铑催化剂[cp*rhcl2]2(1.0mg,0.001mmol),三氟甲磺酰亚胺银(6.0mg,0.008mmol),醋酸铜(20.0mg,0.08mmol),醋酸钠(10.0mg,0.08mmol),1,2-二氯乙烷(dce,1ml),在温度为80℃中反应4小时。反应结束后冷却至室温,经硅藻土抽滤后,浓缩得到粗产物。粗产物用制备的硅胶板进行层析色谱分离,所选展开剂或洗脱剂为石油醚与乙酸乙酯的体积比20:1,得到产物3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸苯酯(1b):1hnmr(400mhz,cdcl3)δ7.54-7.52(m,2h),7.43-7.37(m,2h),7.27(t,j=8.0hz,2h),7.13(t,j=7.2hz,1h),4.52-4.44(m,2h),3.11(dd,j=14.0hz,29.2hz,2h),1.63(s,3h),1.43(t,j=7.2hz,3h).13cnmr(100mhz,cdcl3)δ168.6,168.0,155.8,150.4,132.3,129.4,129.2,127.7,125.6,121.7,121.4,120.9,70.164.0,43.85,25.8,14.4.
本发明实施例的化学转化提供了一种快速构建多取代异喹啉衍生物,该类分子同时包含乙氧基和苯酯基具有进一步构建复杂多苯环荧光稠环分子的潜能。
实施例36-溴-3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸甲酯(1c)
在一个大气压空气氛围下,向15mlschlenk反应管中依次加入4-溴苯甲亚胺酸乙酯化合物2a(46.0mg,0.20mmol),2-丁烯酸甲酯3a(50mg,0.50mmol),三价铑催化剂[cp*rhcl2]2(2.0mg,0.002mmol),三氟甲磺酰亚胺银(6.0mg,0.008mmol),醋酸铜(15.0mg,0.06mmol),醋酸钠(8.0mg,0.06mmol),1,2-二氯乙烷(dce,1ml),在温度为80℃中反应4小时。反应结束后冷却至室温,经硅藻土抽滤后,浓缩得到粗产物。粗产物用制备的硅胶板进行层析色谱分离,所选展开剂或洗脱剂为石油醚与乙酸乙酯的体积比20:1,得到产物6-溴-3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸甲酯(1c):1hnmr(400mhz,cdcl3)δ7.61(d,j=1.2hz,1h),7.47(dd,j=1.2hz,8.0hz,1h),7.33(d,j=8.0hz,1h),4.44-4.39(m,2h),3.53(s,3h),2.88(d,j=14.4hz,1h),2.73(d,j=14.4hz,1h),1.52(s,3h),1.41(t,j=7.2hz,3h).13cnmr(100mhz,cdcl3)δ170.4,167.0,158.3,131.2,131.0,125.2,124.4,122.0,69.9,64.2,51.3,43.2,25.2,14.3.
本发明实施例的化学转化可以兼容卤代烃取代的芳环并且高效参与偶联反应的芳基碳-溴键,从而为多官能团化的异喹啉类衍生物的高效构建、为其在材料领域内的应用提供可能。
实施例46-溴-3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸苯酚酯(1d)
在一个大气压空气氛围下,向15mlschlenk反应管中依次加入苯甲亚胺酸乙酯化合物2b(46.0mg,0.20mmol),2-丁烯酸苯酚酯3b(84mg,0.50mmol),三价铑催化剂[cp*rhcl2]2(2.0mg,0.002mmol),三氟甲磺酰亚胺银(6.0mg,0.006mmol),醋酸铜(12.0mg,0.06mmol),醋酸钠(8.0mg,0.06mmol),1,2-二氯乙烷(dce,1ml),在温度为80℃中反应4小时。反应结束后冷却至室温,经硅藻土抽滤后,浓缩得到粗产物。粗产物用制备的硅胶板进行层析色谱分离,所选展开剂或洗脱剂为石油醚与乙酸乙酯的体积比20:1,得到产物6-溴-3-乙氧基-1-甲基-1h-异吲哚-1-基乙酸苯酚酯(1d):1hnmr(400mhz,cdcl3)δ7.7(d,j=1.6hz,1h),7.51(dd,j=1.2hz,8.0hz,1h),7.36(d,j=8.0hz,1h),7.30(t,j=8.0hz,1h),7.16(t,j=7.6hz,1h),4.49-4.43(m,2h),3.14(d,j=14.4hz,1h),3.06(d,j=14.4hz,1h),2.03(s,1h),1.62(s,3h),1.43(t,j=7.2hz,3h).13cnmr(100mhz,cdcl3)δ168.3,167.3,157.9,150.3,131.3,130.0,129.5,129.3,125.7,125.3,124.5,122.2,121.3,115.5,70.0,64.3,60.32,43.5,25.5,21.0,14.3.
本发明实施例的化学转化可以高效参与偶联反应的芳基碳-溴键及,从而为多官能团化的异喹啉类衍生物的高效构建、为其在材料领域内的应用提供可能。
实施例53-乙氧基-1-甲基-6-硝基-1h-异吲哚-1-基乙酸甲酯(1e)
在一个大气压空气氛围下,向15mlschlenk反应管中依次加入4-硝基苯甲亚胺酸乙酯化合物2c(30.0mg,0.20mmol),2-丁烯酸甲酯3a(20.0mg,0.50mmol),三价铑催化剂[cp*rhcl2]2(2.0mg,0.004mmol),三氟甲磺酰亚胺银(6.0mg,0.008mmol),醋酸铜(10.0mg,0.04mmol),醋酸钠(6.0mg,0.04mmol),1,2-二氯乙烷(dce,2ml),在温度为80℃中反应4小时。反应结束后冷却至室温,经硅藻土抽滤后,浓缩得到粗产物。粗产物用制备的硅胶板进行层析色谱分离,所选展开剂或洗脱剂为石油醚与乙酸乙酯的体积比20:1,得到产物3-乙氧基-1-甲基-6-硝基-1h-异吲哚-1-基乙酸甲酯(1e):1hnmr(400mhz,cdcl3)δ8.30(s,1h),8.27(d,j=8.4hz,1h),7.61(d,j=8.0hz,1h),4.50-4.42(m,2h),3.53(s,3h),3.00(d,j=15.2hz,1h),2.84(d,j=15.2hz,1h),1.58(s,3h),1.44(t,j=7.2hz,3h).13cnmr(100mhz,cdcl3)δ170.1,166.4,157.6,148.7,137.4,123.8,121.4,116.9,70.4,64.6,51.4,42.9,25.2,14.3.
本发明实施例的化学转化可以快速构建硝基取代的异吲哚类分子,而硝基的进一步转化可以对更复杂的分子的构建提供平台。
实施例63-乙氧基-6-碘-1-甲基-1h-异吲哚-1-基乙酸甲酯(1f)
在一个大气压空气氛围下,向15mlschlenk反应管中依次加入4-碘苯甲亚胺酸乙酯化合物2d(30.0mg,0.20mmol),2-丁烯酸甲酯3a(50.0mg,0.50mmol),三价铑催化剂[cp*rh(ch3cn)3cl2]2(2.0mg,0.006mmol),三氟甲磺酰亚胺银(4.0mg,0.006mmol),醋酸铜(12.0mg,0.06mmol),醋酸钠(8.0mg,0.06mmol),1,2-二氯乙烷(dce,1ml),在温度为80℃中反应4小时。反应结束后冷却至室温,经硅藻土抽滤后,浓缩得到粗产物。粗产物用制备的硅胶板进行层析色谱分离,所选展开剂或洗脱剂为石油醚与乙酸乙酯的体积比20:1,得到产物3-乙氧基-6-碘-1-甲基-1h-异吲哚-1-基乙酸甲酯(1f):1hnmr(400mhz,cdcl3)δ7.83(s,1h),7.68(d,j=7.6hz,1h),7.21(d,j=8.0hz,1h),4.45-4.37(m,2h),3.53(s,3h),2.87(d,j=14.8hz,1h),2.73(d,j=14.8hz,1h),1.52(s,3h),1.41(t,j=7.2hz,3h).13cnmr(100mhz,cdcl3)δ170.4,167.2,158.4,136.7,131.8,131.1,122.2,96.5,69.9,64.1,51.3,43.3,25.21,14.3.
本发明实施例的化学转化可以兼容传统的偶联反应中最常用的芳基碘,从而直观证明该转化提供一种与heck反应互补的氧化heck反应。
实施例75-乙氧基-7-甲基-1-甲苯磺酰-1,7-二氢吡咯[3,4-f]7-吲哚基乙酸甲酯(1g)
在一个大气压空气氛围下,向15mlschlenk反应管中依次加入1-对甲苯基-1h-吲哚-5-甲酰亚胺乙酯化合物2e(40.0mg,0.20mmol),2-丁烯酸甲酯3a(50.0mg,0.50mmol),三价铑催化剂[cp*rhcl2]2(1.0mg,0.002mmol),三氟甲磺酰亚胺银(2.0mg,0.004mmol),醋酸铜(12.0mg,0.06mmol),醋酸钠(8.0mg,0.06mmol),1,2-二氯乙烷(dce,2ml),在温度为100℃中反应6小时。反应结束后冷却至室温,经硅藻土抽滤后,浓缩得到粗产物。粗产物用制备的硅胶板进行层析色谱分离,所选展开剂或洗脱剂为石油醚与乙酸乙酯的体积比20:1,得到产物5-乙氧基-7-甲基-1-甲苯磺酰-1,7-二氢吡咯[3,4-f]7-吲哚基乙酸甲酯(1g):1hnmr(400mhz,cdcl3)δ8.10(s,1h),7.77(d,j=8.0hz,2h),7.60(s,1h),7.56(d,j=3.6hz,1h),7.20(d,j=7.6hz,2h),6.87(d,j=3.6hz,1h),4.48-4.39(m,2h),3.54(s,3h),2.99-2.87(m,2h),2.79-2.74(m,2h),2.31(s,3h),1.59(s,3h),1.42(t,j=6.8hz,3h).13cnmr(100mhz,cdcl3)δ170.7,167.3,152.7,145.1,136.0,135.0,130.8,129.8,128.4,127.3,126.8,113.5,109.6,107.2,69.4,63.9,51.3,44.2,38.6,26.0,21.4,14.4.
本发明实施例的化学转化可以兼容优势杂环化合物吲哚,从而为多官能团化的吲哚类衍生物的高效构建、为其在材料领域内的应用提供可能。
以上所述,以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!