一类硅基取代香豆素衍生物及其合成方法与流程
本发明涉及有机分子荧光染料领域,具体涉及一类硅基取代香豆素衍生物及其合成方法。
背景技术:
香豆素是一种苯并吡喃酮类结构的化合物,有良好的生物活性和光物理性质,且具有细胞相容性好毒性小,易修饰等特点,是一类优良的荧光母体。基于香豆素母体的荧光探针不胜枚举,既有用于小分子比如谷胱甘肽、双氧水、苯硫醇等的检测,也有在细胞成像、蛋白检测等方面的应用。但由于香豆素分子发射波长在450nm附近,不利于细胞成像。因此通过对香豆素分子的骨架进行修饰使其波长红移对于其生物学应用具有重要意义。
相关文献报道(chemicalcommunications,2011,47(14):4162-4164;acschemicalbiology,2011,6(6):600-608.)硅基取代的荧光素或罗丹明荧光光谱能发生一个较大的红移,这和si取代后影响了分子的电子结构及轨道性质有关。但是,目前没有文献报道硅基取代的香豆素的合成、性质与应用。所以合成硅基取代的香豆素衍生物具有重要的研究意义和应用价值。
技术实现要素:
本发明的目的是提供一类硅基取代香豆素衍生物及其合成方法,该类衍生物具有较大的stokes位移,在不同的溶剂中有较宽的光谱范围,能有效的对生物体内极性变化进行监测。
实现本发明目的的具体技术方案是:
一类硅基取代香豆素衍生物,特点是所述衍生物具有式i结构:
其中,r为氮甲基、氮乙基、氮氢乙基、氮四元环、氮乙基五元环或氮乙基六元环。所述衍生物具体形式为:
一种上述的硅基取代香豆素衍生物的合成方法,该方法包括以下具体步骤:
当r=氮甲基、氮乙基、氮四元环、氮五元环和氮六元环时,
步骤1:在干燥的氩气保护的密封容器中加入dmf,冰水浴下加入pocl3,在冰水浴下搅拌30-60min,然后加入化合物1,常温下搅拌6-12h,反应完成后,将反应体系倒入冰水中,过滤,洗涤,得到的固体再次用二氯甲烷溶解,加入无水硫酸钠干燥、过滤,旋蒸除去滤液得到化合物2;其中,所述化合物1与pocl3物质的量之比为1:3~4;
步骤2:在干燥的氩气保护的密封容器中加入甲基三苯基碘化磷、无水thf,冰水浴下加入n-buli,此温度下搅拌20-40min,然后逐滴加入化合物2的thf溶液,加完后在室温下搅拌1-2h,反应完成后,过滤并用石油醚冲洗3-5遍,再用食盐水洗滤液,无水na2so4干燥,真空除去溶剂得到化合物3;其中,所述化合物2、甲基三苯基碘化磷与n-buli物质的量之比为:1:1.2~1.3:1.1~1.2;
步骤3:在干燥的氩气保护的密封容器中加入化合物3、无水thf,在-78℃下加入n-buli,此温度下搅拌1-2h,然后加入二甲基丙烯基氯硅烷,缓慢升至室温,搅拌过夜,反应完成后,加水淬灭反应,乙酸乙酯萃取,盐水洗涤有机相,无水na2so4干燥,真空除去溶剂;柱层析法(硅胶、纯石油醚)进一步纯化,得到化合物4;其中,化合物3、n-buli与二甲基烯丙基氯硅烷物质的量之比为1:1.2~1.3:1.3~1.5;
步骤4:在干燥的氩气保护的密封容器中加入化合物4和催化剂grubbsii,加入无水ch2cl2,搅拌4-8h,旋蒸除去溶剂,加入seo2和1,4-二氧六环,在100℃下回流1-2h,反应完成后,硅藻土过滤,旋蒸除去溶剂;柱层析(硅胶、acoet/石油醚)进一步纯化后得到化合物sica、sicb、sicd、sice和sicf;其中,化合物4、grubbsii与seo2的物质的量之比为1:0.05~0.1:1.1~1.2;
当r=氮氢乙基时,该方法包括以下具体步骤:
步骤1:在干燥的氩气保护的密封容器中加入dmf,冰水浴下加入pocl3,在冰水浴下搅拌30-60min,然后加入化合物1c,常温下搅拌6-12h,反应完成后,将反应体系倒入冰水中,过滤,洗涤,得到的固体再次用二氯甲烷溶解,加入无水硫酸钠干燥、过滤,旋蒸除去滤液得到化合物2c;其中,所述化合物1c与pocl3物质的量之比为1:3~4;
步骤2:在干燥的氩气保护的密封容器中加入甲基三苯基碘化磷、无水thf,冰水浴下加入n-buli,此温度下搅拌20-40min,然后逐滴加入化合物2c的thf溶液,加完后在室温下搅拌1-2h,反应完成后,过滤并用石油醚冲洗3-5遍,再用食盐水洗滤液,无水na2so4干燥,真空除去溶剂得到化合物3c;其中,所述化合物2c、甲基三苯基碘化磷与n-buli物质的量之比为:1:1.2~1.3:1.1~1.2;
步骤3:在干燥的氩气保护的密封容器中加入化合物3c、无水thf,在-78℃下加入n-buli,此温度下搅拌1-2h,然后加入二甲基丙烯基氯硅烷,缓慢升至室温,搅拌过夜,反应完成后,加水淬灭反应,乙酸乙酯萃取,盐水洗涤有机相,无水na2so4干燥,真空除去溶剂;柱层析法(硅胶、纯石油醚)进一步纯化,得到化合物4c;其中,化合物3c、n-buli与二甲基烯丙基氯硅烷物质的量之比为1:1.2~1.3:1.3~1.5;
步骤4:在干燥的氩气保护的密封容器中加入化合物4c和催化剂grubbsii,加入无水ch2cl2,搅拌4-8h,旋蒸除去溶剂,加入seo2和1,4-二氧六环,在100℃下回流1-2h,反应完成后,硅藻土过滤,旋蒸除去溶剂;柱层析(硅胶、acoet/石油醚)进一步纯化后得到化合物5c其中,化合物4c、grubbsii与seo2的物质的量之比为1:0.05~0.1:1.1~1.2;
步骤5:在干燥的氩气保护的密封管中加入化合物5c、pd(pph3)4、1,3-二甲基巴比妥酸及无水ch2cl2,体系在常温下反应16-24h,饱和nahco3水溶液猝灭反应后,ch2cl2萃取;饱和nahco3水溶液和盐水洗涤,无水na2so4干燥;柱层析(硅胶,acoet/ch2cl2)对粗产品进行纯化,得到硅基香豆素衍生物sicc;其中,化合物5c、pd(pph3)4与1,3-二甲基巴比妥酸的物质的量之比为:1:0.05~0.1:0.1~0.2;
当r=氮甲基、氮乙基、氮四元环、氮五元环和氮六元环时,具体合成过程如下式所示:
当r=氮氢乙基时,合成过程如下式所示:
本发明的有益效果:本发明能提供一类具有较大stokes位移的硅基取代香豆素衍生物,其在不同溶剂中具有较宽的光谱范围,是一类良好的溶剂致变色香豆素衍生物,利用其荧光成像特点可以用来对生物体内与极性相关的生物活动进行监测,例如对细胞膜成分的检测,细胞凋亡过程的监测以及对脂肪细胞分化过程中脂肪颗粒的识别成像。
附图说明
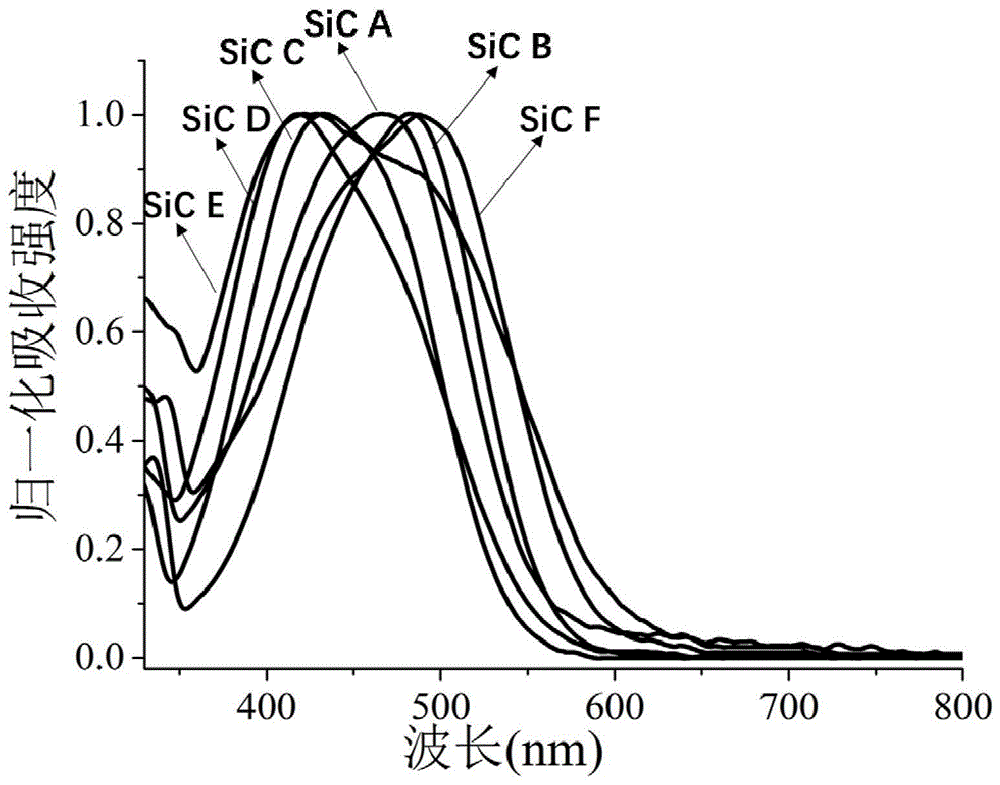
图1为本发明硅基香豆素衍生物在水中的紫外吸收光谱图;
图2为本发明硅基香豆素衍生物在水中的荧光发射光谱图;
图3为本发明硅基香豆素衍生物sicb在不同溶剂中的紫外吸收光谱图;
图4为本发明硅基香豆素衍生物sicb在不同溶剂中的荧光发射光谱图。
具体实施方式
下面用具体实施例对本发明中的硅基香豆素衍生物的合成方法、采用该方法所合成的具有全新结构的硅基香豆素衍生物进一步详细说明。
实施例1
将间溴苯胺(4g,23.26mmol)、3mh2so4(1.8eq,14ml)、甲醛溶液(3eq)加入干燥的烧瓶中,冰浴中搅拌30min,加入nabh4(4eq,3.4g),室温搅拌2小时。反应完成后,加入饱和nahco3溶液,乙酸乙酯萃取。有机相混合,饱和盐水洗2次,无水na2so4干燥,真空除溶剂。柱层析(硅胶、acoet/石油醚)进一步纯化,得到1a(3.95g,85%)。
实施例2
将间溴苯胺(4g,23.26mmol)、碳酸钾(3.16g,1eq)、碘乙烷(8g,2.2eq)、乙腈溶液(40ml)加入干燥烧瓶中,80℃搅拌12小时。反应完成后,用乙酸乙酯过滤萃取。将有机相混合,饱和盐水洗涤,无水na2so4干燥,真空除去溶剂。通过柱层析(硅胶、acoet/石油醚)进一步纯化,得到1b(4.2g,80%)。
实施例3
干燥烧瓶中加入6-溴吲哚啉(2g,10mmol),碳酸钾(1.36g,1eq),碘乙烷(3.43g,2.2eq)和乙腈(20ml),80℃搅拌12小时。反应完成后,过滤,乙酸乙酯萃取。将有机相混合,用饱和盐水洗,用无水na2so4干燥,真空除溶剂。柱层析(硅胶、acoet/石油醚)进一步纯化,得到1e(1.85g,82%)。
实施例4
干燥烧瓶中加入7-溴-1,2,3,4-四氢喹啉盐酸盐(2g,8mmol),碳酸钾(6.66g,6eq),碘乙烷(5g,4eq)和乙腈40ml,80℃下搅拌48小时。反应完成后,过滤后用乙酸乙酯萃取。将有机相混合,饱和盐水洗涤,无水na2so4干燥,真空除溶剂。柱层析(硅胶、acoet/石油醚)进一步纯化,得到1e(1.34g,70%)。
实施例5
r=氮甲基、氮乙基、氮五元环和氮六元环时,
r=氮氢乙基时,
充满氩气的烧瓶加入dmf(10ml)和pocl3(2.8ml,3eq),0℃下搅拌30min。加入化合物1(10mmol)的dmf溶液室温搅拌6h。反应完成后,溶液倒入冰水中,将沉淀物过滤水洗多次后二氯甲烷溶解固体,无水na2so4干燥,旋蒸除去溶剂。柱层析法(硅胶,acoet/)对粗产品进一步纯化,得到2a-2f。产率80%。
化合物2a核磁:1hnmr(500mhz,chloroform-d)δ10.09(s,1h),7.79(d,j=8.9hz,1h),6.79(d,j=2.4hz,1h),6.63(dd,j=8.9,2.0hz,1h),3.08(s,6h).13cnmr(126mhz,chloroform-d)δ190.22,154.49,131.04,129.71,122.01,114.83,110.58,40.09.
化合物2b核磁:1hnmr(500mhz,chloroform-d)δ10.00(s,1h),7.72(d,j=9.0hz,1h),6.72(d,j=2.4hz,1h),6.56(dd,j=9.0,2.1hz,1h),3.36(q,j=7.1hz,4h),1.17(t,j=7.2hz,6h).13cnmr(126mhz,chloroform-d)δ189.75,152.47,130.02,121.44,114.30,110.25,44.77,12.41.
化合物2c核磁:1hnmr(500mhz,chloroform-d)δ10.06(s,1h),7.77(d,j=8.9hz,1h),6.79(d,j=2.4hz,1h),6.62(dd,j=8.9,2.2hz,1h),5.82(ddt,j=16.8,10.2,4.6hz,1h),5.32–5.11(m,2h),3.99–3.95(m,2h),3.44(q,j=7.1hz,2h),1.23(t,j=7.1hz,3h).13cnmr(126mhz,chloroform-d)δ190.02,153.03,131.96,131.21,129.88,122.00,116.84,114.76,110.63,52.47,45.29,12.25.
化合物2e核磁:1hnmr(500mhz,chloroform-d)δ10.04(s,1h),7.57(s,1h),6.47(s,1h),3.62(t,j=8.6hz,2h),3.28(q,j=7.2hz,2h),2.99(t,j=8.4hz,2h),1.21(t,j=7.2hz,3h).13cnmr(126mhz,chloroform-d)δ189.97,157.08,130.37,129.86,124.78,122.63,108.44,51.41,41.06,26.70,11.65.
化合物2f核磁:1hnmr(500mhz,chloroform-d)δ10.02(s,1h),7.52(s,1h),6.68(s,1h),3.44–3.33(m,4h),2.71(t,j=6.3hz,2h),1.94(p,j=6.1hz,2h),1.22(t,j=7.1hz,3h).13cnmr(126mhz,chloroform-d)δ190.03,150.40,129.73,128.02,121.44,121.24,112.81,48.56,45.68,27.42,21.28,11.10。
实施例6
将2-溴-4-氟苯甲醛(2g,8mmol)、碳酸钾(6.66g,6eq)、碘乙烷(5g,4eq)和乙腈(40ml)分别加入干燥的烧瓶中,80℃搅拌48小时。反应完成后,过滤并用乙酸乙酯萃取。将有机相混合,饱和盐水洗涤,无水na2so4干燥,真空除去溶剂。柱层析(硅胶、acoet/石油醚)进一步纯化,得到2d(1.34g,70%)。
化合物2d核磁:1hnmr(500mhz,chloroform-d)δ10.07(s,1h),7.76(d,j=8.6hz,1h),6.46(d,j=2.2hz,1h),6.29(dd,j=8.6,1.8hz,1h),4.03(t,j=7.4hz,4h),2.44(q,j=7.4hz,2h).13cnmr(126mhz,chloroform-d)δ190.20,154.95,131.16,129.58,122.33,113.34,109.21,51.33,16.35。
实施例7
r=氮甲基、氮乙基、氮四元环、氮五元环和氮六元环时
r=氮氢乙基时,
充满氩气的烧瓶中加入甲基三苯基碘化磷(4.8g,1.2eq)和无水thf(20ml),冰浴中搅拌30min。然后逐滴滴加化合物2(10mmol)的thf溶液,室温搅拌1.5h。反应完成后,加入硅胶(200-300目)过滤(石油醚冲洗滤饼3次),再用食盐水洗滤液,无水na2so4干燥,真空除去溶剂。无需进一步纯化。无色油状,产率60%。
化合物3a核磁:1hnmr(500mhz,chloroform-d)δ7.47(d,j=8.8hz,1h),7.01(dd,j=17.4,10.9hz,1h),6.89(d,j=2.5hz,1h),6.67(dd,j=8.8,2.5hz,1h),5.55(d,j=17.4hz,1h),5.16(d,j=10.9hz,1h),2.98(s,6h).13cnmr(126mhz,chloroform-d)δ126.77,124.85,115.53,112.17,111.78,40.31.
化合物3b核磁:1hnmr(500mhz,chloroform-d)δ7.41(d,j=8.8hz,1h),6.96(dd,j=17.4,10.9hz,1h),6.80(d,j=2.7hz,1h),6.59(dd,j=8.8,2.6hz,1h),5.49(d,j=17.4hz,1h),5.09(d,j=10.9hz,1h),3.33(q,j=7.1hz,4h),1.16(t,j=7.1hz,6h).13cnmr(126mhz,chloroform-d)δ135.33,126.94,114.73,111.54,111.16,77.29,44.40,12.53.
化合物3c核磁:1hnmr(500mhz,chloroform-d)δ7.46(d,j=8.8hz,1h),7.02(dd,j=17.4,10.9hz,1h),6.88(d,j=2.4hz,1h),6.65(dd,j=8.8,2.4hz,1h),5.87(ddd,j=16.2,10.3,4.7hz,1h),5.55(d,j=17.4hz,1h),5.22(s,1h),5.19(d,j=5.8hz,1h),5.16(d,j=11.0hz,1h),3.95–3.90(m,2h),3.41(q,j=7.1hz,2h),1.22(t,j=7.1hz,3h).13cnmr(126mhz,chloroform-d)δ148.59,135.33,133.52,126.90,125.02,124.61,116.19,115.11,111.85,111.50,52.50,44.92,12.33.
化合物3d核磁:1hnmr(500mhz,chloroform-d)δ7.40(d,j=8.5hz,1h),6.96(dd,j=17.4,10.9hz,1h),6.57(d,j=2.4hz,1h),6.34(dd,j=8.5,2.3hz,1h),5.50(dd,j=17.4,1.1hz,1h),5.13(dd,j=10.9,1.1hz,1h),3.89–3.85(m,4h),2.37(q,j=7.3hz,2h).13cnmr(126mhz,chloroform-d)δ152.27,135.42,126.81,125.92,124.48,114.59,112.39,110.73,77.05,52.26,16.81。
实施例8
r=氮甲基、氮乙基、氮四元环、氮五元环和氮六元环时,
r=氮氢乙基时,
充满氩气的烧瓶中加入化合物3(5mmol,1.0eq)和无水thf(10ml),-78℃搅拌5min,加入n-buli(1.2eq,3.75ml,1.6m正己烷溶液),-78℃搅拌1h。逐滴加入丙烯基二甲基氯硅烷(0.98ml,1.3eq),搅拌过夜。反应完成后,加水淬灭反应,乙酸乙酯萃取,盐水洗涤有机相,无水na2so4干燥,真空除去溶剂。柱层析法(硅胶、纯石油醚)进一步纯化,得到化合物4,无色油状,产率85%。
化合物4a核磁:1hnmr(500mhz,chloroform-d)δ7.55(d,j=8.6hz,1h),7.01(dd,j=17.1,10.9hz,1h),6.88(s,1h),6.78(d,j=8.5hz,1h),5.84(dq,j=17.3,8.9hz,1h),5.56–5.49(m,1h),5.12(d,j=10.8hz,1h),4.92(dd,j=26.2,13.5hz,2h),2.99(s,6h),1.90(d,j=8.0hz,2h),0.38(s,6h).13cnmr(126mhz,chloroform-d)δ149.18,137.59,135.01,126.07,118.30,113.76,113.50,111.16,40.56,24.34,-1.82.
化合物4b核磁:1hnmr(500mhz,chloroform-d)δ7.54(d,j=8.7hz,1h),7.00(dd,j=17.2,10.8hz,1h),6.84(d,j=2.6hz,1h),6.74(dd,j=8.6,2.4hz,1h),5.92–5.79(m,1h),5.51(d,j=17.2hz,1h),5.09(d,j=11.7hz,1h),4.93(dd,j=25.2,12.7hz,2h),3.43–3.39(m,4h),1.90(d,j=8.1hz,2h),1.22(t,j=7.1hz,6h),0.38(s,6h).13cnmr(126mhz,chloroform-d)δ137.53,135.05,126.21,117.41,113.41,112.87,110.42,44.46,24.31,12.66,-1.86.
化合物4c核磁:1hnmr(500mhz,chloroform-d)δ7.55(d,j=8.7hz,1h),7.02(dd,j=17.2,10.8hz,1h),6.86(d,j=2.6hz,1h),6.76(dd,j=8.7,2.6hz,1h),5.95–5.83(m,2h),5.56–5.50(m,1h),5.25(dd,j=17.2,1.6hz,1h),5.23–5.20(m,1h),5.14–5.09(m,1h),5.00–4.90(m,2h),3.97(d,j=4.9hz,2h),3.48–3.44(m,2h),1.91(d,j=8.1hz,2h),1.24(d,j=7.1hz,3h),0.40(s,6h).13cnmr(126mhz,chloroform-d)δ146.78,137.54,135.05,134.43,127.31,126.11,117.78,115.95,113.43,113.14,110.66,52.80,44.88,24.30,12.43,-1.85.
化合物4d核磁:1hnmr(500mhz,chloroform-d)δ7.53(d,j=8.4hz,1h),7.02(dd,j=17.2,10.8hz,1h),6.59(d,j=2.5hz,1h),6.51(dd,j=8.4,2.4hz,1h),5.85(td,j=17.6,8.1hz,1h),5.54(d,j=17.2hz,1h),5.14(d,j=10.8hz,1h),5.00–4.88(m,2h),3.94(t,j=7.2hz,4h),2.41(p,j=7.2hz,2h),1.90(d,j=8.1hz,2h),0.39(s,6h).13cnmr(126mhz,chloroform-d)δ150.73,137.73,137.37,134.94,132.92,125.85,116.98,113.48,112.46,111.28,52.41,24.24,17.01,-1.90.
化合物4e核磁:1hnmr(500mhz,chloroform-d)δ7.43(s,1h),7.03(dd,j=17.2,10.8hz,1h),6.61(s,1h),5.92–5.81(m,1h),5.53(d,j=17.1hz,1h),5.11(d,j=10.9hz,1h),4.99–4.89(m,2h),3.40(t,j=8.3hz,2h),3.21(q,j=7.2hz,2h),3.01(t,j=8.2hz,2h),1.90(d,j=8.1hz,2h),1.25(t,j=7.2hz,3h),0.39(s,6h).13cnmr(500mhz,chloroform-d)δ151.25,137.89,135.82,135.09,133.57,132.24,121.41,113.37,112.05,110.49,52.10,42.92,28.34,24.43,12.06,-1.72.
化合物4f核磁:1hnmr(500mhz,chloroform-d)δ7.53(d,j=8.4hz,1h),7.02(dd,j=17.2,10.8hz,1h),6.59(d,j=2.5hz,1h),6.51(dd,j=8.4,2.4hz,1h),5.85(td,j=17.6,8.1hz,1h),5.54(d,j=17.2hz,1h),5.14(d,j=10.8hz,1h),5.00–4.88(m,2h),3.94(t,j=7.2hz,4h),2.41(p,j=7.2hz,2h),1.90(d,j=8.1hz,2h),0.39(s,6h).13cnmr(126mhz,chloroform-d)δ150.73,137.73,134.94,125.85,116.98,113.48,112.46,111.28,76.84,52.41,24.24,17.01,-1.90。
实施例9
r=氮甲基、氮乙基、氮四元环、氮五元环和氮六元环时,
r=氮氢乙基时,
充满氩气的烧瓶中加入化合物4(1mmol,1.0eq)、grubbsii催化剂(5%mol)和无水ch2cl2(10ml),混合物在室温下搅拌4h后,真空除去溶剂。将粗产品、seo2(122mg,1.1eq)、dioxane(10ml)加入干燥的烧瓶中,100℃回流1h。反应完成后,硅藻土过滤,旋蒸除去溶剂。柱层析(硅胶、acoet/石油醚)进一步纯化后得到化合物5。橙红色固体,两步产率50%。
化合物sica:1hnmr(500mhz,chloroform-d)δ7.30(d,j=10.9hz,1h),7.24(d,j=8.6hz,1h),6.83(d,j=2.8hz,1h),6.65(dd,j=8.6,2.8hz,1h),5.97(d,j=10.9hz,1h),3.05(s,6h),0.35(s,6h).13cnmr(500mhz,chloroform-d)δ190.24,150.55,148.72,139.68,133.98,129.18,118.52,112.33,76.83,40.10,-4.01.hrms(esi+)calcdfor[m+h]+:232.1152,found:232.1152.
化合物sicb:1hnmr(500mhz,chloroform-d)δ7.29(d,j=10.9hz,1h),7.22(d,j=8.6hz,1h),6.80(s,1h),6.62(d,j=7.3hz,1h),5.95(d,j=10.9hz,1h),3.43(q,j=7.1hz,4h),1.21(t,j=7.1hz,6h),0.35(s,6h).13cnmr(500mhz,chloroform-d)δ148.75,134.27,128.76,126.03,118.08,111.70,76.80,44.44,12.58,-4.06.hrms(esi+)calcdfor[m+h]+:260.1465,found:260.1465.
化合物5c:1hnmr(500mhz,chloroform-d)δ7.29(d,j=10.9hz,1h),7.22(d,j=8.6hz,1h),6.81(d,j=2.5hz,1h),6.64(dd,j=8.6,2.6hz,1h),5.95(d,j=10.9hz,1h),5.85(s,1h),5.17(d,j=22.2hz,2h),3.97(d,j=4.4hz,2h),3.45(q,j=7.1hz,2h),1.22(t,j=7.1hz,3h),0.34(s,6h).13cnmr(500mhz,chloroform-d)δ148.65,134.08,133.18,129.03,126.58,118.51,116.39,112.14,76.78,52.50,44.96,12.37,-4.10.
化合物sicd:1hnmr(500mhz,chloroform-d)δ7.29(d,j=11.0hz,1h),7.21(d,j=8.3hz,1h),6.52(d,j=2.5hz,1h),6.37(dd,j=8.3,2.5hz,1h),5.97(d,j=11.0hz,1h),3.99(t,j=7.3hz,4h),2.43(p,j=7.3hz,2h),0.34(s,6h).13cnmr(500mhz,chloroform-d)δ151.63,148.83,133.81,129.31,117.41,111.27,77.05,51.82,16.66,-4.10.hrms(esi+)calcdfor[m+h]+:244.1152,found:244.1145.
化合物sice:1hnmr(500mhz,chloroform-d)δ7.29–7.24(m,1h),7.06(s,1h),6.56(s,1h),5.94(d,j=11.0hz,1h),3.49(t,j=8.6hz,2h),3.26(q,j=7.2hz,2h),3.00(t,j=9.0hz,2h),1.21(t,j=7.2hz,3h),0.33(s,6h).1hnmr(500mhz,chloroform-d)δ7.29–7.24(m,1h),7.06(s,1h),6.56(s,1h),5.94(d,j=11.0hz,1h),3.49(t,j=8.6hz,2h),3.26(q,j=7.2hz,2h),3.00(t,j=9.0hz,2h),1.21(t,j=7.2hz,3h),0.33(s,6h).13cnmr(500mhz,chloroform-d)δ153.10,149.13,139.04,129.12,128.83,128.24,112.36,77.30,51.34,41.91,27.76,11.88,-4.06.hrms(esi+)calcdfor[m+h]+:258.1309,found:258.1308.
化合物sicf:1hnmr(500mhz,chloroform-d)δ7.25(d,j=10.9hz,1h),6.97(s,1h),6.73(s,1h),5.95(d,j=10.9hz,1h),3.44(q,j=7.1hz,2h),3.40–3.33(m,2h),2.76(t,j=6.3hz,2h),1.98(dt,j=11.6,6.1hz,2h),1.20(t,j=7.1hz,3h),0.36(s,6h).13cnmr(126mhz,chloroform-d)δ148.87,146.05,137.55,133.70,128.75,126.17,122.98,116.92,48.77,45.33,27.96,21.72,11.16,-3.98.hrms(esi+)calcdfor[m+h]+:272.1465,found:272.1463。
实施例10
充满氩气的反应管中加入化合物5c(100mg,0.37mmol)、pd(pph3)4(21mg,5%mol)和1,3-二甲基巴比妥酸(68mg,1.2eq)。然后加入无水ch2cl2(2ml),室温搅拌24h。饱和nahco3水溶液猝灭反应后,ch2cl2萃取。饱和nahco3水溶液和盐水洗涤,无水na2so4干燥。柱层析(硅胶,acoet/ch2cl2)对粗产品进行纯化,得到sicc(55mg,65%)。
化合物sicc:1hnmr(500mhz,chloroform-d)δ7.29(d,j=11.0hz,1h),7.21(d,j=8.4hz,1h),6.74(d,j=2.6hz,1h),6.56(dd,j=8.4,2.6hz,1h),5.96(d,j=10.9hz,1h),3.32(s,1h),3.23(q,j=7.2hz,2h),1.29(t,j=7.2hz,3h),0.33(s,6h).13cnmr(500mhz,chloroform-d)δ149.19,148.87,134.41,129.39,128.04,120.16,112.44,77.16,38.14,14.75,-4.04.hrms(esi+)calcdfor[m+h]+:232.1152,found:232.1155。
化合物光学性质的分析:
1.溶液配制
分别精确称取化合物sica、sicb、sicc、sicd、sice、sicf1-2mg,用dmso溶解并配成1mm的高标溶液,备用。
2.样品吸收光谱的测定
将配好的样品溶液用紫外分光光度计(shimadzuuv-2600)(石英皿,光路长度1cm)测定其紫外吸收光谱,测定范围300-800nm,测定其最大吸收波长,计算其样品的摩尔吸光系数,结果见表1,吸收光谱图见附图1。并选取sicb,测定其在不同溶剂中的紫外吸收光谱,结果见表2,吸收光谱图见附图3。
3.样品发射光谱的测定
将配好的样品溶液用荧光光谱仪(shimadzurf-6000)测定其荧光发射光谱,激发波长488nm,测定范围520-800nm,测定其最大发射波长,结果见下表1,样品的荧光发射光谱见附图2。并选取sicb,测定其在不同溶剂中的荧光发射光谱,结果见表2,荧光发射光谱图见附图4。
表1.化合物光谱数据
表2.化合物光谱数据
- 还没有人留言评论。精彩留言会获得点赞!