一种利用多重定量PCR技术检测样品中多种病毒的方法与流程
本发明涉及生物检测领域,特别是涉及一种利用多重定量pcr技术检测样品中多种病毒的方法。
背景技术:
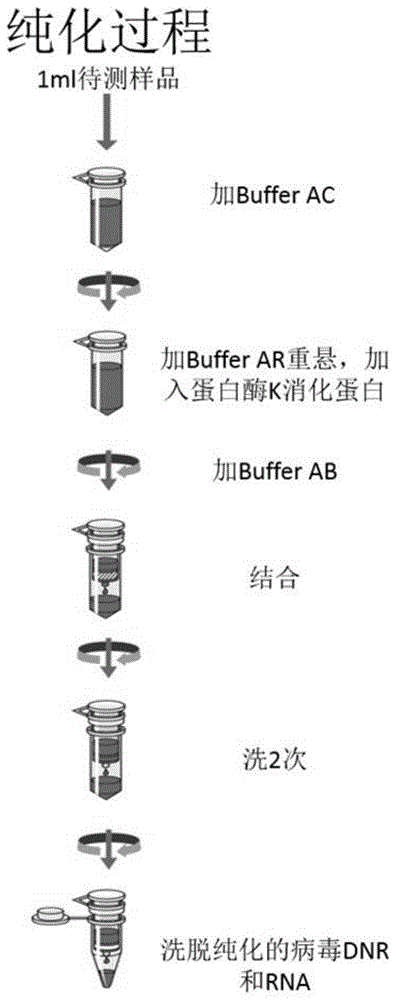
:2018年各国监管机构共计批准上市了39个生物药。2018年全球销量前十的药物中,包括8个生物药,销售金额近700亿美元。大部分生物药(如单克隆抗体)以注射方式应用于疾病治疗,这些生物药的质量与患者的用药安全密切相关。各国监管机构出台了一系列法规来确保生物药的生物安全性,如法规中明确要求对生物制品的下游纯化工艺去除或灭活病毒的能力进行研究,即对相关纯化工艺步骤进行病毒清除验证。目前很多重组蛋白类生物制品以中国仓鼠卵巢细胞(cho)作为表达宿主,cho细胞自身就存在内源性逆转录病毒样颗粒,虽然理论上该病毒样颗粒对人类不具有致病性,但仍有潜在的风险。根据国内外法规,异嗜性小鼠白血病病毒(x-mulv)可以作为逆转录病毒样颗粒的模型病毒,用于生物制品下游纯化工艺的病毒清除研究。伪狂犬病毒(prv),是一种引起牛、羊、猪、犬、猫等多种家畜和野生动物发热、奇痒及脑脊髓炎为主要症状的疱疹病毒,常作为疱疹病毒的模型病毒用于病毒清除研究。在病毒清除实验中,为了检测纯化步骤对模型病毒的去除效果,通常使用tcid50或病毒斑块实验对病毒进行滴度检测。这些基于指示细胞的病毒滴度检测需要首先进行毒性实验和干扰实验等预实验。借助预实验结果,细胞分析可以呈现测试样品中有活力的病毒颗粒或病毒感染性。对病毒或病毒颗粒的计数受到指示细胞状态和培养时间的影响,这会导致不同实验人员或不同实验间的差异。在接种含有病毒的测试样品后,通常需要花费5-10天的时间才能获得数据,而包括预实验在内的最终实验数据的获得则需要2-3个月的时间。聚合酶链式反应(pcr)是检测目标核酸片断的有力工具,pcr通过热循环驱动扩增。从1981年发明该技术以来,多种改进程序被引入以满足分子生物学领域的科研需求。定量pcr(qpcr)被广泛用于细胞中mrna的定量以及检测目标蛋白的表达水平。该技术已被开发应用于病毒检测以及病毒清除研究。qpcr主要的方法学是通过实时监测荧光强度来检测pcr产物的含量。而在常规pcr中,扩增产物通过电泳进行分离和可视化。qpcr中使用的dna聚合酶具有5’至3’的核酸内切酶活性,通过水解标记有荧光素和淬灭染料的寡核苷酸探针,产生荧光信号。将qpcr和荧光素标记探针相结合,理论上可以检测单个病毒dna/rna,在实际应用中qpcr可以检测到样品中个位数的病毒dna/rna,无论病毒的活性如何。目前,将qpcr用于病毒清除验证的例子是检测proteina亲和柱工艺流程中单一种类的包膜病毒,因为目标产物的洗脱液是低ph缓冲液,可以灭活包膜病毒,因此基于细胞学测试无法检测到任何病毒感染性。在病毒清除研究中,通常只添加一种病毒对某个工艺步骤进行验证。但在实际生产中生物制品有同时被多种病毒污染的风险,故有必要对层析和纳滤等工艺去除多种病毒的能力进行验证。由于生物制品对不同病毒及其指示细胞的干扰程度不一致,且一种病毒往往具有侵染多种指示细胞并形成斑块的能力,所以基于细胞的检测方法不适用于同时检测样品中的多种病毒。因此,开发一种基于pcr技术快速检测样品中多种病毒的方法显得十分必要。技术实现要素:本发明所要解决的技术问题在于,提供一种基于pcr技术检测样品中多种病毒的方法,以克服现有方法不适用于同时检测样品中的多种病毒的缺陷。为解决以上技术问题,本发明提供以下技术方案:一种利用多重定量pcr技术检测样品中多种病毒的方法,包括以下步骤:(1)通过待检测病毒dna和/或rna序列进行blast比对,设计并优化针对多种待检测病毒的特异性引物及探针;(2)利用步骤(1)的特异性引物和探针来优化多重qpcr的反应条件,获得了优选的反应条件;(3)通过稀释病毒原液和经典的细胞斑块实验,将103或104pfu的病毒稀释液调整为103或104拷贝的标准化ct值;根据调整后稀释液和稀释因子,从高滴度病毒库纯化制备106拷贝的病毒核酸标准品;将106拷贝病毒核酸标准品按10倍梯度稀释产生1到105拷贝的目标病毒基因标准品,进而通过abi7500pcr仪器检测产生病毒的标准曲线;(4)采用一步法taqman检测试剂盒,按照所述步骤(2)得到的优选的反应条件对进行检测;根据步骤(3)的标准曲线计算检测结果。在一种具体的实施方式中,所述步骤(1)中,待测病毒为x-mulv病毒和prv病毒。在一种具体的实施方式中,针对x-mulv病毒和prv病毒,所述特异性引物为:x-mulv病毒的正向引物(fp1),其核苷酸序列为gtgcaggagcctcggtacaa,如seqidno.1所示;x-mulv病毒的逆向引物(rp1),其核苷酸序列为tcgatcattaggttggtaactctccaag,如seqidno.2所示;prv病毒的正向引物(fp2),其核苷酸序列为gacggaaaagtacctgctcatg,如seqidno.3所示;prv病毒的逆向引物(rp2),其核苷酸序列为tctgcggaggtacgagatgga,如seqidno.4所示;所述探针为:x-mulv病毒的探针(probe1),其核苷酸序列为tcgacagccctcaccaggtcttcaatg,如seqidno.5所示;prv病毒的探针(probe2),其核苷酸序列为gtgacagccctcaccaggtcttcaat,如seqidno.6所示。在一种具体的实施方式中,所述步骤(2)中优选的反应条件为:1)扩增体系:1-stepmastermix终浓度1x,fp-1终浓度900nm,rp-1终浓度900nm,probe-1终浓度200nm,fp-2终浓度900nm,rp-2终浓度900nm,probe-2终浓度200nm,模板,10μl,用ddh2o补足25μl体系;2)扩增参数:step1:48℃,30min;step2:95℃,10min;step3:95℃,15sec;step4:60℃,1min;step3和step4,40cycles;在一种具体的实施方式中,所述步骤(4)中,将所述x-mulv病毒的rna先反转录成dna,再和prv病毒的dna一起检测。在病毒检测中,需要按照法规要求,定量或定性检测生物制品中的病毒。将qpcr技术应用于病毒定量的主要障碍包括:1)产生假阳性信号,而不是假阴性信号;2)用于测定病毒基因组拷贝数的合适的标准曲线;3)利用获得的基因拷贝数,计算病毒滴度的方法;4)在一个检测样品中加入多种病毒。本发明以x-mulv和prv为例,设计了通过多重定量qpcr方法同时检测样品中多种模型病毒(dna病毒和rna病毒)滴度的方法。本发明设计并优化后的x-mulv和prv引物和探针,具有高特异性和灵敏性,分别进行单基因扩增,扩增曲线呈现明显的指数期、平台期。本发明利用x-mulv和prv病毒原液的滴度和经典的病毒斑块实验,将病毒原液稀释并制备成106核酸标准品。使用标准品进行梯度稀释,制成1到105拷贝数的目标病毒基因标准品,进而生成标准曲线。使用该标准品生成的标准曲线,线性关系和相关系数好,孔间差异小,重复性高。本发明中x-mulv病毒核酸为rna,prv病毒为dna。x-mulv病毒需要先反转录成dna,才能和prv的dna一起检测。本发明利用多重qpcr定量检测x-mulv病毒的rna和prv病毒的dna。该方法在线性标准曲线斜率、线性相关系数、准确度、精密度、特异性、检测限等方面,都符合ich法规要求。本发明的方法使用经优化设计的引物和探针、qpcrmix反应体系以及扩增参数,结合病毒核酸标准曲线标定,对样品中不同种类的模型病毒进行精确定量检测,并可以用于生物制品病毒清除研究的方法。附图说明图1为本发明中病毒核酸纯化示意图。图2为本发明中prv单基因扩增标准曲线和扩增图谱。图3为本发明中prv双基因扩增标准曲线和扩增图谱。图4为本发明中x-mulv单基因扩增标准曲线和扩增图谱。图5为本发明中x-mulv双基因扩增标准曲线和扩增图谱。具体实施方式下面将对本发明的技术方案进行清楚、完整的描述,显然,所描述的实施例是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例一1.在emem培养基中分别加入不同体积比的病毒,以产生不同的供试品(sn-004,sn-005)。使用商业化病毒核酸纯化试剂盒纯化核酸,具体纯化步骤见附图1:2.使用设计优化后的x-mul和prv病毒的引物和探针,优化后的qpcr反应条件进行多重和单重qpcr检测对比,具体步骤如下:1)待测试样品:sn-004、sn-005、sn-007注:sn-004和sn-005分别为5ul测试体积。sn-007为sn-0045ul+sn-0055ul,10ul测试体积。2)配制qpcrmix配制:单基因qpcrmix配制:1-stepmastermix终浓度1x,fp终浓度900nm,rp终浓度900nm,probe终浓度200nm,模板5μl,用ddh2o补足25μl体系。双基因qpcrmix配制:1-stepmastermix终浓度1x,fp-1终浓度900nm,rp-1终浓度900nm,probe-1终浓度200nm,fp-2终浓度900nm,rp-2终浓度900nm,probe-2终浓度200nm,模板,10μl,用ddh2o补足25μl体系。3)abi7500qpcr仪运行参数:step148℃,30min,step295℃,10min,step395℃,15sec,step460℃,1min,step3&step4,40cycles。4)qpcr结果:标准曲线和扩增图片见附图2~5,x-mulv和prv病毒检测拷贝数见表1和表2。表1各基因定量检测结果表2单基因和多基因定量检测结果比较平均值(单基因)平均值(双基因)平均值标准偏差值rsd%prv132.678177.311154.99531.56020.362x-mulv364.551346.651355.60112.6573.5593.结论:1)从附图2~5中x-mulv和prv的标准曲线的斜率和r2可以看出,通过本技术方案制备的标准品进行qpcr检测的扩增效率在90%-105%,结果可信。2)从附图2~5中x-mulv和prv的扩增图谱可以看出,扩增曲线光滑,有明显的指数扩增期、平台期;另外标准曲线1拷贝可以检出。说明本技术方案设计的引物探针的特异性和敏感性都很高,该qpcr检测所用的扩增体系、扩增参数和该引物探针的适应性高。从qpcr结果可以看出,x-mulv和prv的单基因和多基因定量结果差异不大,rsd<50%,可以使用多重qpcr定量检测x-mulv和prv病毒的dna/rna。综上所述,上述各实施例仅为本发明的较佳实施例而已,并不用以限定本发明的保护范围,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,皆应包含在本发明的保护范围内。序列表<110>苏州药明检测检验有限责任公司<120>一种利用多重定量pcr技术检测样品中多种病毒的方法<130>cpc-np-19-101754<160>6<170>siposequencelisting1.0<210>1<211>20<212>dna<213>正向引物(正向引物)<400>1gtgcaggagcctcggtacaa20<210>2<211>28<212>dna<213>逆向引物(逆向引物)<400>2tcgatcattaggttggtaactctccaag28<210>3<211>22<212>dna<213>正向引物(正向引物)<400>3gacggaaaagtacctgctcatg22<210>4<211>21<212>dna<213>逆向引物(逆向引物)<400>4tctgcggaggtacgagatgga21<210>5<211>27<212>dna<213>探针(探针)<400>5tcgacagccctcaccaggtcttcaatg27<210>6<211>26<212>dna<213>探针(探针)<400>6gtgacagccctcaccaggtcttcaat26当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1