液滴单细胞表观基因组谱分析用于患者分层的用途
本发明属于分子生物学、抗药性和微流体领域。特别地,本发明涉及一种测定微流体液滴中的核酸以用于抗药性的诊断和/或预后和患者分层的方法。
背景技术:
真核生物基因组被组织化成染色质,其使得能够不仅压缩dna,而且还调节dna代谢(复制、转录、修复、重组)。因此,当前的挑战是理解(i)功能性染色质结构域是如何在细胞核中建立的,(ii)染色质结构/信息是如何通过组装、拆卸、修饰和重塑机制而动态化的,和(iii)这些事件是如何在疾病建立以及疾病的进展和复发中参与/维持的。理解这些事件将允许确定疾病进展的新机制和新的治疗靶标,以及治疗性分子的控制作用。
此外,生物学中的基本研究问题是了解数百种不同的细胞类型是如何从多细胞生物中的相同遗传物质产生的。许多不同细胞类型不能仅通过遗传学来解释,而是通过可以将表型与基因型联系起来的额外信息来解释。1942年,康拉德·h·沃丁顿(conradh.waddington)创造了表观遗传学一词以指“研究基因及其产物之间因果相互作用的生物学分支,其使表型形成”。“表观遗传信息”的这一额外层次储存为构成染色质的dna和组蛋白两者的化学修饰的形式。表观遗传机制通过染色质修饰来调节基因表达和塑造特定的染色质景观(landscape),其允许进行对细胞类型和组织身份的预测。
dna和组蛋白修饰通过充当能够读取信息的效应蛋白的识别位点和通过稳定其与染色质的结合来参与到各种基于dna的过程之中。高丰度的组蛋白修饰使得能够严格控制染色质结构且在对基于dna的过程的调节中具有极大灵活性。这种多样性导致了可在不同位点处同时修饰的组蛋白之间的串扰(wang等,2008,naturegenetics40(7):897-903)。
组蛋白修饰可以正面地或负面地影响彼此。此外,组蛋白修饰之间的交流也与其他染色质修饰(例如dna甲基化)一同存在,其均参与微调生物学功能的整体调控(du等,2015,naturereviewsmolecularcellbiology,16(9):519-532)。
dna和组蛋白修饰有助于定义在不同染色质状态内的表观基因组特征,所述表观基因组特征对细胞类型和组织身份具有高指示性。可以利用对这些标记的全基因组谱分析来了解基因组调控的全局景观,然后例如区分正常细胞和疾病细胞状态的背景下的表观基因组差异(consortiumepigenomics2015,nature518(7539):317-329)。然而,染色质谱分析技术的当前状态不允许研究细胞异质性,也不允许检测染色质状态中的细胞间差异。
使用传统的chip-seq方法对表观遗传修饰、表观遗传标记物/擦除物(eraser)、在染色质结构中起作用的因子、2d和3d组织化的全基因组作图需要大量细胞以生成高质量的结合位点谱分析。多项研究已显示优化的chip-seq方案将输入材料从数百万个细胞减少至数百个细胞,而不丢失在对富集或耗竭区域的检测中的分辨率(adli等,2010,naturemethods7(8):615-618;brind’amour等,2015,naturecommunications6:6033;ma等,2018,scienceadvances4(4):eaar8187)。然而,这些方法仅产生修饰状态的平均快照,而无法提供对表观遗传异质性的见解。
在单细胞分辨率下对组蛋白修饰进行谱分析仍然具有挑战性,部分是因为免疫沉淀过程中与非特异性结合相关联的噪声水平倾向于随低量起始原料增加。从一个单一细胞免疫沉淀染色质在技术上是可行的,但会导致高度可变的结果。
可以预先用特定且独特的dna序列(条形码)将来自分离的单细胞的染色质索引化,然后与来自数个至数千个细胞的索引化染色质结合,以成批地进行免疫沉淀,如传统chip-seq方案中。这种方法在保留单细胞信息的同时,规避了与低输入材料的免疫沉淀中的高实验噪声相关的问题。的确是,对于一个细胞特有的条形码,每个读数(read)可在测序后归属于其原始细胞。然而,就像其他涉及分子索引化的单细胞技术一样,只有索引化核小体具有被扩增和测序的可能性。
在这方面,rotem开发了drop-chip技术,该技术将染色质索引化方法与基于液滴的微流体技术相结合以对数千个细胞的组蛋白修饰进行谱分析(rotem等,2015,naturebiotechnol.33(11):1165-1172)。该液滴形式为进行单细胞分析提供了一种多功能的工具。在通过液滴中的细胞的微球菌核酸酶进行区室化(compartmentalization)、裂解和染色质片段化的步骤之后,然后将所述液滴与包含dna条形码的第二群体的液滴一对一融合,允许在单细胞水平上对染色质进行索引化。
尽管使用drop-chip揭示了在胚胎干细胞的群体中的不同染色质状态,但由于低染色质索引化效率或索引化核小体的不良回收,单细胞信息仅限于每细胞检测到的少至数百个独特的富集化基因座。值得注意的是,drop-chip技术受到两个主要限制,其可负面地影响每细胞回收的信息量。首先,只有对称索引化的核小体才可以被扩增并可以成为测序文库的一部分。这个要求极大地增加了系统的严格性,并对核小体(即仅两端均连接至条形码的那些)施加了强力的选择。其次,索引化核小体的扩增仅依赖于聚合酶链反应(pcr)的众多循环,其增加了引入扩增偏倚和错误的可能性。
未经处理的细胞中的染色质状态的自发异质性可以是获得抗药性的关键分子组成部分,不管癌症治疗的作用机制是什么。许多类型的癌症最初易受化学治疗药物的影响,随时间可通过这些和其他机制产生抗药性。然而,抗药性的方法可能是疾病特异性的,而其他方法可以是进化保守的。化学疗法和靶向疗法的抗性的出现是癌症治疗的主要挑战。在未经治疗的肿瘤内的遗传异质性现被认为是抗药性的关键决定因素。此外,预计非遗传且特别是转录和表观遗传机制在面对环境、代谢或治疗相关压力的癌细胞的适应中发挥作用(rathert,p.等,nature525,543-547,(2015);kim,c.等,cell173,879-893e813,(2018))。通过组蛋白修饰调节染色质结构是主要的表观遗传机制和基因表达的调节因素,然而,染色质特征对肿瘤异质性和进化的贡献仍然是未知的。考虑到影响本领域已知方法的上述局限,显然需要用于与不同染色质状态相关的抗药性的诊断和/或预后的改进的方法和组合物,其在微流体液滴中使用单细胞表观遗传谱分析。
本发明的某些实施方式公开了在未经治疗的、药物敏感的肿瘤中的罕见细胞群体显示出与抗性细胞的染色质特征相匹配的染色质特征。本发明人已经开发了液滴微流体方法以在单细胞分辨率下以高达10,000个基因座/细胞的覆盖率对数千个细胞的染色质景观进行谱分析,该方法具有如本文所公开的多种应用。
技术实现要素:
本发明的一个方面涉及一种用于抗药性的诊断和/或预后的组合物和方法,其中通过使用微流体系统对从受试者获得的细胞中的单细胞染色质状态进行谱分析,所述方法包括以下步骤:
a.提供至少第一类型的液滴,其中所述第一类型的液滴包含
i.生物要素,
ii.裂解缓冲液,和
iii.核酸酶,
b.在暂时使所述核酸酶失活的条件下收集所述第一类型的液滴,
c.孵育所述第一类型的液滴,从而重新激活所述核酸酶,
d.提供至少第二类型的液滴,其中所述第二类型的液滴包含核酸序列,
e.融合所述第一类型的液滴和所述第二类型的液滴,从而生成第三类型的液滴,
f.孵育所述第三类型的液滴,从而将所述核酸序列连接至一个或多个感兴趣的基因组区域,
g.对所述一个或多个感兴趣的基因组区域进行测序。
所述一个或多个感兴趣的基因组区域是修饰的基因组区域,所述修饰的基因组区域包含核酸序列和/或与核酸序列相关联的蛋白质复合物。
所述修饰的基因组区域包含翻译后修饰,所述翻译后修饰选自乙酰化、酰胺化、脱酰胺化、羧化、二硫键、甲酰化、糖基化、羟基化、甲基化、肉豆蔻酰基化、亚硝基化、磷酸化、异戊烯化、核糖基化、硫酸化、sumo化、泛素化及其衍生物,优选甲基化。
根据本发明,所述细胞源自处于疾病状态和/或怀疑处于疾病状态的受试者或健康受试者。所述受试者处于疾病状态,并且所述疾病状态包括癌症、传染病、自身免疫疾病、代谢疾病、炎症疾病、遗传和非遗传疾病。
根据本发明所述的方法,其中所述疾病状态是乳腺癌。
根据本发明所述的方法,其中所述细胞是未经治疗的和/或经治疗的。所述经治疗的细胞是用治疗剂或化学治疗剂或化学药物或生物药物来治疗的。
根据本发明所述的方法,其中所述经治疗的细胞是用他莫昔芬和卡培他滨来治疗的。
根据本发明所述的方法,其中所述单细胞染色质状态已经丢失了促进抗药性的基因的染色质标记,其中所述染色质标记是组蛋白修饰h3k4me3和h3k27me3。
附图说明
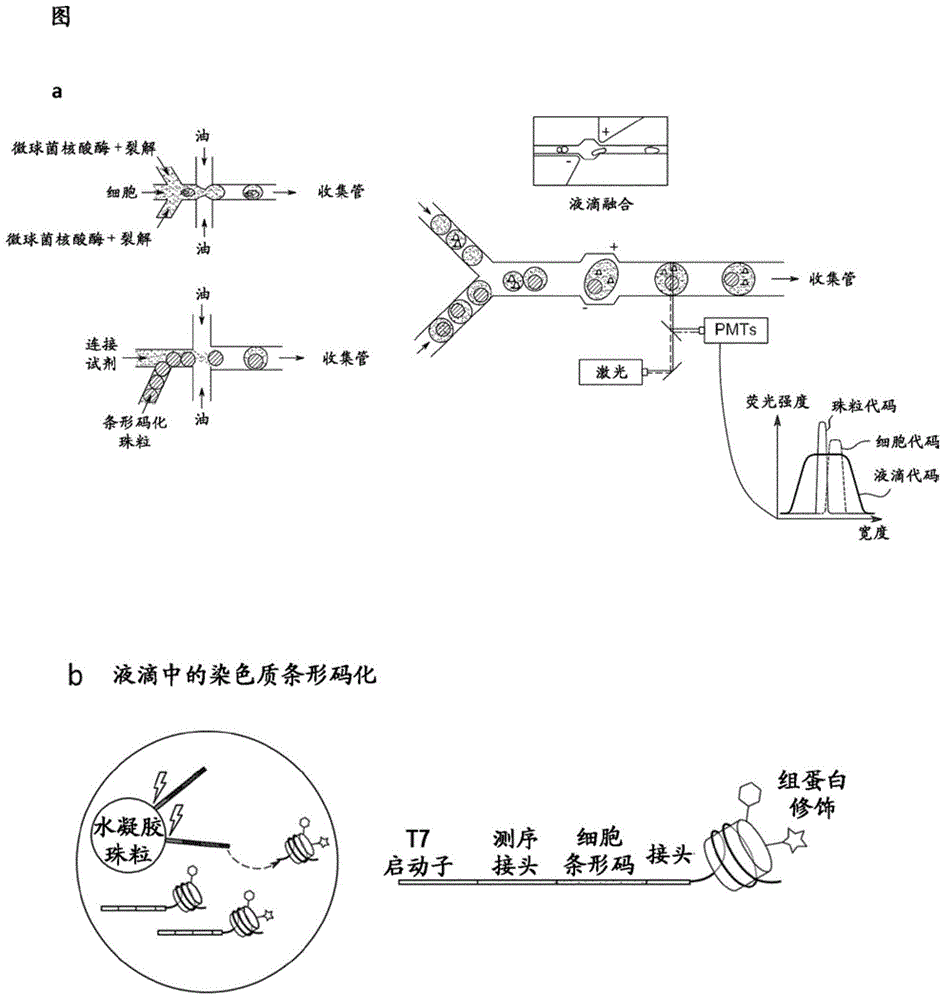
图1描绘了根据本发明的微流体工作流程。(a)在45pl液滴中用裂解和染色质片段化所需的试剂将细胞区室化。平行地,将带有dna条形码的水凝胶珠粒与连接试剂一起包封于100pl液滴中。在融合设备中重新注入两种乳液,将条形码液滴(100p1)和核小体液滴(45p1)非对称地配对,并且电场触发融合。用激光束对融合液滴进行一对一扫描并实时分析每个液滴的组成。(b)收集融合液滴的乳液以在液滴中进行核小体条形码化。通过光切割从珠粒释放条形码并将其连接至核小体。合并液滴的内容物,进行免疫沉淀,并对富集的dna进行测序。与条形码相关的读数的反卷积(deconvolution)将所有序列归属于其原始细胞以重建单细胞染色质谱分析。
图2描绘了液滴之间的微球菌核酸酶(mnase)活性的同步和暂停。特别地,图2a显示了在微球菌核酸酶孵育的不同时间点的来自人jurkatt细胞的dna片段的凝胶电泳。在t=0分钟时,dna尚未片段化,这证实了微球菌核酸酶活性在冰上液滴收集之后同步。时间点t=12分钟+1小时冰显示了与12分钟孵育后相似的消化谱分析,这证实了微球菌核酸酶活性在液滴被存储于冰上时暂停。图2b描绘了从固定细胞核开始的液滴中微球菌核酸酶活性的暂停。微球菌核酸酶暂停3小时,而没有染色质过度消化。
图3描绘了使液滴中微球菌核酸酶活性完全失活所必需的egta浓度。通过tapestation测量孵育之后剩余的寡核苷酸的分数,并相对于消化阴性对照(即不含微球菌核酸酶的液滴)进行标准化。26mm最终浓度的egta使液滴中的微球菌核酸酶完全失活。柱状图显示了重复实验的未消化寡核苷酸的平均分数和对应于标准差的误差条。
图4描绘了根据本发明的核酸序列。新结构(v2)通过在条形码的两端上均添加pac1限制性位点的一半来允许消化条形码多联体(concatemer)。还添加了下链的3’端处的c3间隔子以强制执行该连接的方向。用保护基使非全长条形码完整,所述保护基包含修饰的碱基修饰,所述修饰的碱基修饰防止不需要的连接。该表描述了每个条形码结构在测序之后识别的正确条形码的比例。
图5描绘了条形码化水凝胶珠粒的质量控制。(a)在从水凝胶珠粒进行光切割之后的dna条形码的tapestation谱分析显示了全长条形码的存在(在146bp处的较大的峰),以及未能完整的中间体(在72bp、94bp和119bp处的峰)。(b)使用辐射荧光显微镜对液滴中的水凝胶珠粒进行成像。从左至右:(i)明场图像;(ii)将与lllumina测序接头互补的dna探针杂交至条形码上之后的成像;(iii)在通过光切割释放液滴中的条形码之后,与(ii)相同。比例尺是35μm。(c)单珠粒深测序结果,其显示了16个珠粒的前两个最丰富的条形码的分数。平均而言,97.7%的存在于珠粒之上的条形码与同一序列匹配,而第二丰富的条形码仅代表所有测序读数的0.17%。
图6描绘了对液滴中连接效率的估计。该连接进行2小时、4小时和过夜孵育。两种测量方法均给出了对连接产物的分数的相似估计,并且在过夜孵育后观察到效率的显著提高(~10%)。柱状图显示了重复实验(在不同的两天进行的实验)的连接寡核苷酸的平均分数,误差条对应于标准差。
图7描绘了概念验证研究和预期结果。(a)人b和t淋巴细胞被分别在液滴中包封并特定索引化。然后将来自两种乳液的索引化染色质组合以用于靶向3种不同组蛋白修饰(h3k4me3、h3k27ac和h3k27me3)的chip-seq。(b)数据分析会使用无监督聚类方法将细胞成组为两个聚类。然后会通过细胞类型特异性条形码序列来确认基于b和t细胞的染色质谱分析的正确聚类。
图8描绘了对液滴中的细胞和水凝胶包封的监控。(a)针对在1.8khz下分析的液滴记录的实验时间追踪。橙色荧光存在于所有液滴中,用于控制液滴的大小(液滴代码)。绿色荧光指示液滴中细胞的存在(细胞代码)。细胞代码强度(绿色)相对于每个液滴中的液滴代码强度(橙色)的图。包含细胞的液滴具有高绿色细胞代码强度,从而允许以高于噪声水平的细胞门(cell-gate)的定义对包封的细胞进行计数。调节为λ=0.1的细胞密度导致了~9%的包含一个单一细胞的液滴。(b)针对在650hz下分析的100pl液滴记录的实验时间追踪。橙色荧光存在于所有液滴中,用作液滴代码以控制液滴大小。红色荧光指示液滴中水凝胶珠粒的存在(珠粒代码)。珠粒代码强度(红色)相对于每个液滴中的液滴代码强度(橙色)的图。通过使用珠粒的紧密堆积排序,65%至75%的液滴包含一个珠粒。
图9描绘了对液滴融合的实时监控。(a)针对在150hz下融合后的液滴记录的实验时间追踪。橙色荧光存在于所有液滴中,用作液滴代码以控制融合液滴的大小。绿色荧光指示细胞的存在,红色荧光指示水凝胶珠粒的存在。蓝色荧光是细胞乳液特定的液滴代码。(b)液滴代码“细胞”强度相对于每个液滴中的液滴代码强度的图,其定义了融合之后的液滴的4个主要群体。中间的主要群体代表正确配对并融合的液滴(70%至80%)。来自珠粒乳液的未配对的液滴在下方群体中,而来自具有高蓝色荧光强度的细胞乳液的未配对液滴在左上方群体中。最后一个群体(右上)与包含与1个珠粒液滴融合的2个细胞液滴的错误配对的液滴相关联。(c)绘制细胞代码强度相对于每个液滴中的珠粒代码强度的图,使得能够精确计数可用液滴(包含一个细胞和一个珠粒的那些)。来自小图(a)中的时间追踪的液滴被指示为不同群体的实例。
图10描绘了细胞总数以及在h3k4me3和h3k27me3单细胞chip-seq实验中的微流体站上通过荧光检测到的与条形码化水凝胶珠粒共包封的细胞的数量。测序数据分析显示了与在微流体站上计数的包含细胞和珠粒两者的液滴的数量紧密相关的识别的条形码的数量,表明该系统具有高的整体效率。
图11描绘了scchip-seq程序识别亚群体的灵敏度。代表在检测限的计算机模拟中的h3k27me3scchip-seq数据集的t-sne图,其具有t细胞群体中掺入的b细胞的变化比率(从上至下)和每条形码的独特映射读数的变化阈值(从左至右)。根据细胞类型特异性条形码序列对点进行着色。
图12描述了在drop-chip中的测序性能相对于本发明人的程序。表1比较了在dropchip中和在本发明人的scchip-seq系统中的每测序文库的预期细胞的数量、原始测序读数的数量,以及每细胞的原始读数的平均数量。表2比较了在测序之后识别的细胞的数量,在qc之后的分析中使用的最终细胞数量,和在dropchip中和在本发明人的scchip-seq系统中的qc之后的每细胞的可用读数的平均数量。
图13描绘了人和小鼠细胞混合物确认了单细胞分辨率。(a)与小鼠相对于人类参比基因组校准的每条形码读数数量的散点图,其显示96.5%的条形码对一个物种具有特异性(至少95%的读数具有与两个物种之一映射的相同条形码)。小鼠(26.4%)、人(70.1%)和混合(3.5%)物种的百分比接近于基于液滴中细胞的泊松分布的预期值,每液滴的平均细胞数λ为0.1(分别为32.6%、65.2%和2.2%)。(b)柱状图,其显示了相比于在微流体站上计数的预期细胞数(灰色条;从包含1/3小鼠细胞和2/3人细胞的混合物中总计3,000),对每个物种(蓝色、红色和黑色条)识别的条形码数(对应于细胞数)。
图14描绘了单细胞chip-seq数据的聚类揭示了细胞类型特异性生物相似性。(a)h3k4me3和h3k27me3单细胞chip-seq数据集中每条形码(即每细胞)的scchip-seq原始和独特测序读数的分布的直方图。(b)密度散点图,其代表了并行收集且处理以生成h3k4me3单细胞chip-seq数据集的b细胞的相同乳液的三个独立分数之间的log2累积计数。重复实验之间的相关性是基于跨单细胞的5kb基因组分组(genomicbin)中每百万读数的累积计数来计算的。全基因组计算皮尔森相关性得分(pearson’scorrelationscore)和p值。(c)两个生物重复实验的密度散点图,对应于从不同细胞培养瓶中收集并用不同批次的条形码化水凝胶珠粒处理以产生h3k27me3单细胞chip-seq数据集的b细胞的两种乳液。重复实验之间的相关性是基于跨单细胞的50kb基因组分组中每百万读数的累积计数来计算的。全基因组计算皮尔森相关系数和p值。(d)t-sne图,其代表来自两个生物重复实验的h3k27me3scchip-seq数据,根据原始批次(左)或一致性聚类结果(右)着色,反对细胞群体聚类的批次效应。(e)左图:来自1:1人b细胞和t细胞混合群体以及分别条形码化的b细胞和t细胞的h3k27me3scchip-seq数据的细胞对细胞皮尔森相关系数的层次聚类和对应热图。独特的读数计数、原始批次和一致性聚类结果显示于热图上方。右图:对应的t-sne图,根据细胞类型特异性条形码序列,来自该混合群体的点着色为灰色,而来自分别条形码化的b细胞和t细胞的点着色为不同的灰色阴影。(f)维恩图(venndiagram),其比较了t细胞和b细胞数据集通过单细胞和批量方法检测到的h3k4me3峰。
图15描绘了从单细胞chip-seq谱分析重建细胞类型特异性染色质状态。(a)t-sne图,其代表了来自人b和t淋巴细胞的h3k4me3和h3k27me3scchip-seq数据集,所述人b和t淋巴细胞通过使用携带单细胞和细胞类型特异性条形码两者的水凝胶珠粒而在液滴中分别索引化,混合用于免疫沉淀。根据细胞类型特异性条形码序列,点为不同的灰色阴影。准确率代表通过scchip-seq数据的一致性聚类进行的分类(图16a)和通过细胞类型特异性条形码评估的已知细胞身份之间的一致性。(b)差异富集基因座的快照(图16b),其具有针对每个细胞类型的累积单细胞谱分析和批量谱分析。由威尔科克森符号秩检验(wilcoxonsigned-ranktest)识别的差异结合区域以灰色表示,带有相应调节的p值和log2倍数变化。(c)散点图,其显示了累积单细胞相对于批量chip-seq数据中的log2rpm(每百万映射读数的读数计数)富集,对于h3k4me3在5kb基因组分组内计算,对于h3k27me3在50kb基因组分组内计算。全基因组计算皮尔森相关系数和p值。
图16描绘了单细胞chip-seq数据将人t细胞(jurkat)与人b细胞(ramos)区分开。(a)h3k4me3(上图)和h3k27me3(下图)scchip-seq数据集的一致性聚类矩阵。一致性得分范围为0(白色:从不聚类在一起)至1(深蓝色:总是聚类在一起)。(b)火山图,其代表差异分析的经调节p值(威尔科克森符号秩检验)相对于倍数变化,所述差异分析比较了针对h3k4me3(上图)和h3k27me3(下图)scchip-seq数据集的b细胞和t细胞之间的染色质特征(0.01的q值阈值和1的|log2fc|阈值)。(c)柱状图,其显示了来自h3k4me3scchip-seq数据集中的通路分析的经调节p值的-log10。柱状图下方显示了前10个重要的基因集。
图17描绘了获得的针对卡培他滨的抗性的乳腺肿瘤模型。(a)用卡培他滨治疗n=6只具有hbcx-95衍生物的小鼠,持续6周。图代表随时间变化的相对肿瘤体积(rtv,mm3)。当pdx的体积达到超过200mm3(#35、#40!小鼠)时,对患有复发性肿瘤的小鼠进行第二轮卡培他滨治疗。#40小鼠对卡培他滨无反应,以及提取1080mm3的pdx标本并标记为hbcx-95-capar。手稿中研究的样品用虚线矩形表示。(b)未经治疗的hbcx-95和卡培他滨抗性hbcx-95-caparpdx中每细胞的scchip-seq原始和独特测序读数的分布的直方图。(c)散点图,显示累积单细胞相对于成批h3k27me3chip-seq数据中的log2富集,其是基于50kb基因组分组中每百万映射读数的计数计算的。全基因组计算皮尔森相关性得分和p值。(d)在累积单细胞和成批h3k27me3chip-seq数据的50kb基因组分组中的log2富集的分布。每个chip-seq方法(成批或单细胞)的右上角显示最大和最小log2富集之间的幅度。
图18描绘了小鼠基质细胞的单细胞chip-seq谱分析,揭示了来自h3k27me3染色质标记的细胞身份。(a)方案,其代表了获得性化学疗法抗性的pdx模型的产生(图17a)和实验。对于hbcx-95,样品颜色代码为深灰色,对于hbcx-95-capar,样品颜色代码为浅灰色。(b)scchip-seq数据集的t-sne示意图,其根据起源肿瘤(卡培他滨敏感性或抗性,左图)或一致性聚类结果以不同的灰色阴影表示(图19g)。(c)scrna-seq数据集的t-sne示意图,其根据起源肿瘤或一致性聚类结果着色(图21b)。对每个亚群体指示通过差异表达分析识别的标记基因(图21c)。(d-e)左图:在来自聚类chrom_c2(ptk2)或chrom_c3(lrmp)的细胞中显著耗尽h3k27me3的基因组区域相比于其他细胞的快照,以及每种细胞类型和簇的累积单细胞谱分析。每个簇内具有h3k27me3富集的细胞的数量和百分比显示于轨迹上方。通过威尔科克森符号秩检验识别的差异结合区域以灰色高亮显示,并带有相应调节的p值和log2倍数变化,参比簇用星号表示。中图:scchip-seq数据集的t-sne示意图,根据ptk2和lrmp基因座的每个细胞中的h3k27me3富集信号对点进行着色。右图:scrna-seq数据集的t-sne示意图,根据ptk2和lrmp在每个细胞中的表达信号,点为不同的灰色阴影。
图19描绘了对来自hbcx-95模型的基质细胞的scchip-seqh3k27me3谱分析的详细分析。(a)具有每个细胞的独特映射读数的不同最小数量的pca图,显示h3k27me3scchipseq主要成分是主要受高达每细胞1,600个读数的细胞覆盖率驱动。(b)细胞间皮尔森相关性得分的层次聚类和对应热图,其基于其针对n=949个小鼠基质细胞的h3k27me3谱分析,最小覆盖为1,600个独特映射读数。热图上方显示了来自层次聚类的聚类成员资格、原始样品(hbcx-95为绿色,hbcx-95-capar为粉红色)和独特读数计数。(c)通过层次聚类识别的每个聚类的簇内相关性得分的分布。(d)实际scchip-seq数据集(浅灰色)和随机数据集(黑色曲线)的细胞间皮尔森相关性得分的分布。上百分位数由一条垂直的黑线表示。(e)仅针对“相关”细胞的层级聚类和相应细胞间皮尔森相关性得分热图,代表所有细胞的81.8%。(f)左:针对范围为2至10的k个簇,绘制了簇的成员之间的所有成对相关性得分的均值。右:范围为2至10的k个簇的平均簇内相关性得分。在k=3个簇时,簇内相关性最大化。(g)scchip-seq小鼠基质细胞的层次聚类和相应细胞间一致性聚类得分热图。一致性得分的范围是0(白色:从不聚类在一起)到1(深蓝色:总是聚类在一起)。簇成员资格在热图上方显示为不同灰色阴影。
图20描绘了来自hbcx-95模型的小鼠基质细胞的单细胞chip-seqh3k27me3谱分析的监控分析。(a)左图:来自每个簇的细胞中的差异结合区域相比于通过威尔科克森符号秩检验识别的所有剩余细胞。如果经调节p值低于0.01并且倍数变化大于1,则认为基因组区域是富集的(深灰色)或耗尽的(浅灰色)。中图和右图:条形图显示通路分析中经调整p值的-log10(使用msigdb,q值<0.1),所述通路分析是针对h3k27me3中特别是chrom_c2和chrom_c3中耗尽的区域。前十个重要的基因集显示在条形图上。(b)左图:来自chrom_c2(col4al)的细胞相对于所有其他细胞的差异耗竭基因座的快照,具有每种细胞类型和簇的累积单细胞图谱。由威尔科克森符号秩检验识别的差异结合区域以灰色表示,具有相应调节的p值和log2倍数变化,相对于chrom_c2计算。每个簇内具有h3k27me3富集的细胞的数量和比例在轨道上方显示。右图:scchip-seq和scrna-seq数据集的t-sne表示,根据col4al基因座的每个细胞中的h3k27me3富集和表达信号对点进行着色。
图21描绘了来自hbcx-95模型的小鼠基质细胞的单细胞rna-seq图谱的聚类和监控分析。(a)细胞间皮尔森相关性得分的层次聚类和相应热图。热图上方显示了来自一致性聚类的簇成员资格、原始样品(hbcx-95为绿色,hbcx-95-capar为粉红色)和独特读数计数。(b)基质细胞的层次聚类的一致性聚类得分。一致性得分的范围是0(浅灰色:从不聚类在一起)到1(深灰色:始终聚类在一起)。簇成员资格在树状图上用颜色表示。(c)t-sne图,根据每个细胞中代表每个细胞群体的每个标记基因的表达信号对点进行着色。
图22描绘了来自hbcx-95模型的人肿瘤细胞的单细胞chip-seqh3k27me3谱分析和scrna-seq谱分析的聚类。(a)左图:卡培他滨抗性pdx(hbcx-95-capar)相对于未经治疗的pdx(hbcx-95)中0.5mb非重叠区域的拷贝数的图,从成批chip-seq实验的输入中获得。中图和右图:以灰色表示的卡培他滨抗性pdx和未经处理的pdx的成批dna谱分析的受拷贝数变化影响的基因座快照。(b)左图:细胞间皮尔森相关性得分的层次聚类和相应热图。热图上方显示了原始样品(hbcx-95为深灰色,hbcx-95-capar为浅灰色)和独特读数计数。中图:scrna-seq肿瘤细胞的t-sne图,根据原始样品和一致性聚类分割,点为不同的灰色阴影。右图:scrna-seq肿瘤细胞的层次聚类的一致性聚类得分。一致性得分的范围是0(白色:从不聚类在一起)到1(深蓝色:总是聚类在一起)。树状图下方的聚类成员资格为不同的灰色阴影。(c)scchip-seq数据集的一致性聚类分析。左图:对范围为2至10的k个簇,绘制了簇的成员之间所有成对相关性得分的平均值。在k=2个簇时,簇内相关性最大化。右图:肿瘤细胞(hbcx-95和hbcx-95-caparpdx)上scchip-seq的细胞间一致性聚类得分的层次聚类和对应热图。一致性得分的范围是0(浅灰色:从不聚类在一起)到1(深灰色:总是聚类在一起)。热图上方的簇成员资格为不同灰色阴影。(d)条形图,显示了来自抗性细胞中具有h3k27me3耗尽的区域的通路分析的经调节p值的-log10。基因集在条形图上显示。(e)左图:对于图23f中识别的基因座,显示了chrom_c1和chrom_c2的聚集的h3k27me3染色质谱分析,显著差异地富集和表达。对于用灰色指示的每个窗口,指示了每个簇内的log2倍数变化,经调节p值(q值),具有h3k27me3富集的细胞的数量和比例。右图:scrna-seq数据集的t-sne示意图。根据每个细胞中的表达信号,点为不同的灰色阴影。
图23描绘了用卡培他滨治疗的三阴性乳腺癌的pdx模型中敏感且具抗药性的特定的h3k27me3染色质景观。(a)scchip-seq数据集的细胞间皮尔森相关性得分的层次聚类和对应热图。对于hbcx-95,样品颜色代码为深灰色,对于hbcx-95-capar,样品颜色代码为浅灰色,并且独特读数计数在热图上方显示。(b-c)scchip-seq数据集的t-sne示意图,根据原始样品(b)和一致性聚类分割(c),细胞为不同的灰色阴影(图22c)。(d)关于chrom_c2的项目一致性得分,分数1对应于高度代表chrom_c2簇的细胞。虚线表示相对于chrom_c2(左线)或chrom_c1(右线)的项目一致性得分为0.9,深灰色细胞源自hbcx-95,粉红色细胞源自hbcx-95-capar。三角形突出显示了具有超过0.9的一致性得分并且与它们的原始样品相反的细胞。(e)代表经调节p值的火山图(威尔科克森符号秩检验)与log2倍数变化的差异分析,其用于比较chrom_c2和c1之间的染色质富集(阈值q值为0.01,|log2fc|为l)。(f)左图:饼图,表示与tss重叠且可检测的转录的差异富集窗口的数量。右图:来自hbcx-95-capar和hbcx-95的细胞之间的log2表达倍数变化,其针对差异富集基因座中所有检测到的基因(n=37)。条形图根据log2fc和相关的q值进行着色(黑色表示q>0.01,绿色表示表达不足,红色表示表达过度)。(g-h)左图:显示了igf2bp3和col4a2每个簇的h3k27me3染色质图谱。每个簇内具有h3k27me3富集的细胞的数量和比率显示在轨迹上方。右图:代表scrna-seq数据集的t-sne图,根据igf2bp3或col4a2的细胞表达信号,点为不同的灰色阴影。(i)在chrom_c2中的h3k27me3中耗竭的hoxd基因座(但scrna-seq没有可检测的转录)的聚集的h3k27me3染色质图谱。
图24描绘了在用他莫昔芬处理的管腔er+pdx模型中,来自敏感性肿瘤的部分细胞具有抗性细胞的h3k27me3染色质特征。(a)scchip-seq数据集的细胞间皮尔森相关性得分的层次聚类和对应热图。对于hbcx-22,原始样品用深灰色表示,对于hbcx-22-tamr,用浅灰色表示,独特读数计数在热图上方显示。(b)scchip-seq数据集的t-sne示意图,根据原始样品(左)和一致性聚类分割(右)以不同灰色阴影着色细胞(图25c-d)。(c)关于chrom_c2的项目一致性得分。得分1对应于高度代表chrom_c2群集的细胞。虚线表示相对于chrom_c2(上线)或chrom_cl(下线)的项目一致性得分为0.9。(d)饼图表示与tss重叠且可检测到的转录的显著差异富集(h3k27me3,q<0.01)的窗口数。(e)scrna-seq数据集的细胞间皮尔森相关性得分的层次聚类和对应热图。hbcx-22的原始样品显示为绿色,hbcx-22-tamr的原始样品显示为粉红色,热图上方显示了umi计数。(f)scrna-seq数据集的t-sne示意图,根据原始样品(左)和一致性聚类分割(右)以不同的灰色阴影着色细胞(图25f)。(g)hbcx-22-tamr与hbcx-22相比,前10条上调通路(具有最低q值)中每条通路的平均表达得分的层次聚类。在热图上方标出了原始样品、rna簇和独特读数计数。(h-i)左图:每个簇的聚集h3k27me3染色质图谱的egfr和igfbp3基因座的快照。对于每个窗口,均显示log2倍数变化和经调节p值。中图:条形图显示每个簇中具有h3k27me3富集的细胞的比率。对应的细胞数显示在条形图上方。对于每个簇,细胞的来源在下方显示(hbcx-22为深灰色,hbcx-22-tamr为浅灰色)。右图:条形图显示每个簇中的细胞与所有剩余细胞的egfr和igfbp3表达水平的平均log2倍数变化。条形图上方显示了每个簇中可检测到的egfr或igfbp3表达的细胞比率。对于每个簇,细胞的来源在下方显示(hbcx-22为深灰色,hbcx-22-tamr为浅灰色)。
图25显示了来自hbcx-22模型的人肿瘤细胞的单细胞chip-seq图谱的聚类情况。(a)未经治疗的pdx(hbcx-22)和抗他莫昔芬的hbcx-22-tamrpdx中的每个细胞的scchip-seq原始和独特测序读数的分布的直方图。(b)针对抗他莫昔芬的pdx(hbcx-22-tamr)与未经治疗的pdx(hbcx-22)的整体dna图谱绘制的0.5mb非重叠区域中的拷贝数。在该异种移植模型中未发现拷贝数的异常变化。(c)scchip-seq数据集的一致性聚类分析。左图:针对范围为2至10的k个簇,绘制了簇成员之间所有成对相关性得分的平均值。在k=2个簇时,簇内相关性最大化。右图:肿瘤细胞(hbcx-22和hbcx-22-tamrpdxs)上的scchip-seq的细胞间一致性聚类得分的层次聚类和对应热图。一致性得分的范围是0(浅灰色:从不聚类在一起)到1(深灰色:总是聚类在一起)。位于热图上方的簇成员为不同灰色阴影。(d)代表经调节p值(威尔科克森等级检验)与倍数变化的火山图,用于差异分析,比较了chrom_c2和chrom_c1之间的染色质标记(q值<0.01和|log2fc|>1)。(e)条形图,其显示了从在来自chrom_c2的细胞中耗竭h3k27me3的区域的通路分析得到的经调节p值的-log10。基因集在条形图中显示。(f)scrna-seq肿瘤细胞(fibcx-22和fibcx-22-tamrpdxs)的细胞间一致性聚类得分的层次聚类和对应的热图。一致性得分的范围是0(浅灰色:从不聚类在一起)到1(深蓝色:总是聚类在一起)。簇成员资格在热图上方用颜色编码。(g)左图:针对alcam基因座,显示了chrom_c1和chrom_c2的聚集的fi3k27me3染色质图谱。对于显示为灰色的每个窗口,显示了log2倍数变化和经调节p值。中图:条形图,其显示每个簇中具有fi3k27me3富集的细胞比率。条形图上方显示了相应的细胞数。每个簇的细胞来源显示于下方(fibcx-22为深灰色,fibcx-22-tamr为浅灰色)。右图:条形图,其显示每个簇中的细胞与所有剩余细胞的alcam表达水平的平均log2倍数变化。条形图上方显示了每个簇中具有可检测到的alcam表达的细胞的比率。每个簇的细胞来源显示于下方(fibcx-22为深灰色,fibcx-22-tamr为浅灰色)。
具体实施方式
本发明人已经开发了一种基于液滴微流体的改进的单细胞chip方法,所述方法相比于在rotem中公开的drop-chip技术导致了每个个体细胞富集的基因座的数量的5至10倍增加(参见图7)。该方法允许以高灵敏度和高精度在单细胞水平评估组蛋白修饰、dna修饰的碱基(包括用于在单细胞或任何生物要素水平识别正在进行的dna复制事件的修饰的核苷酸)、染色质/dna相关因子。该方法适用于识别具有与另一者不同的特征的细胞群或任何生物要素,这些特征是组蛋白和/或dna修饰、因子的存在。这些要素的存在或不存在然后潜在地指示基因表达中的变化,因此可以用作生物标记物、恢复该等变化的治疗靶标。
充分理解的是,细胞可以表示细胞核,作为染色质结构的区室。细胞或细胞核或任何生物要素可以是固定的生物要素。固定剂的实例包括醛(包括但不限于甲醛、多聚甲醛),醇(包括但不限于乙醇和甲醇),氧化剂,汞,苦味酸盐,羟乙基哌嗪乙硫磺酸(hepes)-谷氨酸缓冲液介导的有机溶剂保护作用(hope)固定剂。
如在drop-chip中,分别生成包含细胞的液滴和包含条形码的液滴,然后在专用的微流体融合设备中将其重新注入并一对一融合(见图1)。然而,本发明的方法在表征条形码策略的至少两个方面不同于rotem。第一,本发明人用携带数百万个独特或最丰富的dna序列的水凝胶珠粒(或任何固体载体)代替了从包含寡核苷酸的微量滴定板乳化的可溶性条形码。第二,根据本发明的一个方面的条形码结构的新型设计允许线性扩增所有条形码化核小体,而非仅如rotem中那样在两端上均对称地条形码化的核小体。第三,条形码设计包括增加感兴趣的核酸的条形码化的效率的额外特征。这些特征包括将保护性部分添加至“不完整条形码”以防止它们附着于感兴趣的核酸。在另一个方面,条形码可包含在全长寡核苷酸上的保护性碱基(保护性碱基包括但不限于硫代磷酸酯(phosphorotioate)、lna/bna、核苷酸亚磷酰胺(phosphoramitidite)、合成环、非3’oh或5’p碱基、2’-o-甲基-dna/rna),这些保护性碱基将保护全长条形码,而非全长条形码可用核酸外切酶来消化。
可以添加一组额外的条形码(称为“实验条形码”)以在单个免疫沉淀反应中多路进行不同实验。后续生物信息学分析将允许基于“实验条形码”的序列对实验条件进行多路分解。
充分理解的是,条形码是可将源自一个区室的核酸的具体特征与源自另一区室的核酸的具体特征区分开的核酸序列。这些条形码的产生是本领域技术人员已知的,并且可以代表随机序列(侧接或不侧接已知序列),或通过拆分合并合成(splitpoolsynthesis)产生(klein等,cell,2015)。
此外,根据本发明的一个方面的方法的特征在于同步/暂停步骤,所述同步/暂停步骤限制了液滴之间的染色质消化中的细胞间变化。
前述优点公开于下文表征本发明的一个方面的方面和实施方式中。在实施例和图中提供了本发明的实施。
在本发明的一个方面,提供了一种使用微流体系统来识别一个或多个感兴趣的基因组区域的方法,所述方法包括以下步骤:
a.提供至少第一类型的液滴,其中所述第一类型的液滴包含
i.生物要素,
ii.裂解缓冲液,和
iii.核酸酶,
b.在暂时使所述核酸酶失活的条件下收集所述第一类型的液滴,
c.孵育所述第一类型的液滴,从而重新激活所述核酸酶,
d.提供至少第二类型的液滴,其中所述第二类型的液滴包含核酸序列,
e.融合所述第一类型的液滴和所述第二类型的液滴,从而生成第三类型的液滴,
f.孵育所述第三类型的液滴,从而将所述核酸序列连接至至少一个感兴趣的基因组区域,
g.对所述一个或多个感兴趣的基因组区域进行测序。
本发明的方法是在微流体系统中实现的。在本发明的一个方面的背景下,术语“微流体系统”是指具有通常以微米或亚微米规模制造的一个或多个通道和/或腔室的系统或装置。
本发明的方法的特征在于存在第一、第二和第三类型的液滴。如本文所使用的,与液滴相关联的术语“第一”、“第二”和“第三”用于根据液滴的内容物来区分液滴。由于该方法是在微流体系统中进行的,因此术语“液滴”也指“微流体液滴”。因此,在微流体系统的背景下,术语“液滴”也指被第二流体包围的第一流体的单独部分,其中第一流体和第二流体是不混溶的。
根据本发明的方法的阶段或步骤,液滴可包含于微流体系统中(芯片上)或与微流体系统分开的收集器设备中(芯片外)。液滴可具有球形或非球形形式。
在本发明的一个实施方式中,液滴的体积范围为约20pl至约100pl。优选地,液滴的体积范围为约30pl至约70pl。更优选地,液滴的体积范围为约40pl至约50pl。理想地,液滴的体积为约45pl。如本文所用,术语“约”是指规定值的值±10%的范围。
如本文所用,术语“裂解缓冲液”是指能够裂解生物细胞的缓冲液。术语“裂解缓冲液”的含义在本领域技术人员的公知常识之内。
如本文所用,术语“基因组区域”是指dna或rna编码的核酸序列。
如本文所用,术语“核酸酶”是指能够切割连接核酸分子中的核苷酸残基的磷酸二酯键的酶试剂。核酸酶可消化双链、单链、环式和线性核酸分子。在本发明的背景下,核酸酶可以是切割在多核苷酸链内的磷酸二酯键的核酸内切酶,或切割在多核苷酸链的末端处的磷酸二酯键的核酸外切酶,可以是转座酶。核酸酶也可以是位点特异性核酸酶,其切割在特定核苷酸序列(例如识别序列)内的特定磷酸二酯键。核酸酶的非限制性实例是微球菌核酸酶(micrococcalnuclease,mnase)。在一个特定的实施方式中,核酸酶是微球菌核酸酶(mnase)。
如本文所用,术语“生物要素”可指单个细胞、细胞核、包含核酸的细胞器(例如线粒体),并且可以从生物体、人或非人受试者获得。在后一种情况下,非人受试者不限于哺乳动物受试者。
由于细胞是以不同时间尺度依次处理的,因此对液滴中的单个生物要素进行酶促测定具有挑战性。例如,细胞或任何生物要素的包封步骤持续约20分钟,其是与孵育步骤相同的数量级。因此,在一开始包封于液滴中的细胞或任何生物要素与生产结束时包封于液滴中的细胞或任何生物要素相比将与核酸酶接触更长的时间。对于融合设备中液滴的重新注入,可以作出类似的观察(参见图1中的一般方案)。确实,取决于实验的设计,两种乳液的融合可持续1小时至4小时,这意味着一些含有片段化dna的液滴在融合之前以及其微球菌核酸酶被egta失活之前“等待”数小时。因此,同步和暂停酶活性是至关重要的,以避免引入在个体细胞或任何生物要素之间的染色质消化变化。
值得注意的是,在常规批量chip-seq分析中,核酸酶的失活在加入egta的核酸酶孵育后立即发生。不同的是,在单细胞chip-seq分析中,egta不可以立即加至液滴内,并且核酸酶仅在与包含条形码的液滴融合后才被失活。
为了控制和限制染色质消化中细胞间或任何生物要素变化,本发明人引入了在使所述核酸酶暂时失活的条件下收集第一类型的液滴的步骤。目的是使液滴中的核酸酶活性同步/暂停的所述收集步骤是在每个孵育步骤之前进行的。本发明人发现,液滴区室使微球菌核酸酶对温度变化敏感且能够选择性地阻断/重新激活并重新阻断酶活性。对核酸酶活性的这种严格控制在批量中是不可能的。该影响被怀疑不是依赖于单独的微球菌核酸酶活性,而是依赖于任何酶。
因此,根据另一个实施方式,所述方法进一步包括在步骤(e)之前在使所述核酸酶暂时失活的条件下收集第一类型的液滴的步骤。
在又一个实施方式中,步骤(b)的条件包括选择-20℃至10℃范围的温度,并且步骤(c)的条件包括选择20℃至40℃范围的温度。
可以在微流体系统外(芯片外)孵育液滴以进行单细胞染色质片段化。当裂解发生在液滴中时,来自裂解细胞的核dna可接触核酸酶。因此,消化的动力学对于优先地产生单核小体特别重要,所述单核小体保留于液滴中。
在一个特定的实施方式中,定时所述孵育步骤(c)以获得片段化为单核小体的核dna。
在又一个实施方式中,所述一个或多个感兴趣的基因组区域是修饰的基因组区域。
根据本发明,所述修饰的基因组区域包含与核酸序列相关联的蛋白质复合物和/或核酸序列。在一个特定的实施方式中,所述修饰的基因组区域是修饰的单核小体。在另一个实施方式中,所述修饰的基因组区域是转录因子结合位点、染色质修饰子结合位点、染色质重塑子(remodeler)位点、组蛋白伴侣(chaperone)结合位点。
根据本发明,所述修饰的基因组区域还可包含翻译后修饰,所述翻译后修饰选自乙酰化、酰胺化、脱酰胺化、羧化、二硫键、甲酰化、糖基化、羟基化、甲基化、肉豆蔻酰基化、亚硝基化、琥珀酰化(assuccinylation)、丁酰化、磷酸化、异戊烯化、核糖基化、硫酸化、sumo化(sumoylation)、泛素化及其衍生物。
根据本发明,修饰的基因组区域还可包含组蛋白变体,所述组蛋白变体选自cenp-a/cid/cse4(着丝粒的表观遗传标记物)、h3.3(转录)、h2a.z/h2av(转录/双链断裂修复)、h2a.x(性染色体的双链断裂修复/减数分裂重塑)、macroh2a(基因沉默/x染色体失活)、h2a.bbd(活性染色质的表观遗传标记物)、h3.z(对外部刺激的细胞应答的调节)、h3.y(对外部刺激的细胞应答的调节)。
根据本发明,所述修饰的基因组区域包含修饰的dna序列,所述修饰的dna序列选自甲基化及其衍生物、修饰的核苷酸(例如edu、brdu、idu、cldu等)。最常见的修饰碱基的方法是添加甲基标记物,并且在各种物种中,已经在胞嘧啶和腺嘌呤上发现甲基化,产生5mc、n4-甲基胞嘧啶(n4mc)或6-甲基腺嘌呤(6ma)、5-羟甲基胞嘧啶(5hmc)、5-甲酰基胞嘧啶(5fc)和5-羧基胞嘧啶(5cac)。
如上文介绍的,在rotem中公开的方法的显著局限在于,只有对称索引化的核小体才可被扩增,并且可为测序文库的一部分。该要求极大地增加了系统的严格性,并对核小体施加了强力的选择,其限于两端均与条形码连接的那些。与drop-chip方法相反,本发明人已经出人意料地发现,从仅一端对核小体进行索引化会增加单细胞覆盖率并最终提高该系统在单细胞染色质谱分析之间区分更细微变化的能力。
在又一个实施方式中,核酸序列不对称地连接至所述至少一个或多个感兴趣的基因组区域。
如本文所用,术语“不对称连接”是指与感兴趣的基因组区域连接的至少一个条形码的存在,由此所述连接是仅与所述感兴趣的基因组区域的两个末端之一。
在本发明进一步的方面,提供一种核酸序列,其包含:
a.至少一个索引序列,
b.测序接头,和
c.至少一个位于3’-和/或5’-端处的保护官能团。
如本文所用,术语“核酸序列”是指单链或双链核酸。在另一个实施方式中,“核酸序列”可以是dna或rna。在一个优选的实施方式中,“核酸序列”是双链dna。在一些实施方式中,“核酸序列”包含双链dna,所述双链dna包含第一链条形码和第二链条形码。在一些实施方式中,所述第一链条形码和第二链条形码包含互补序列。在一些实施方式中,所述第一链条形码和第二链条形码包含非互补序列。
如本文所用,术语“索引序列”是指独特的核苷酸序列,所述独特的核苷酸序列可与任何其他索引序列以及核酸序列(其中其被包含)内的任何其他核苷酸序列区别开来。“索引序列”可以是随机的或特别设计的核苷酸序列。“索引序列”可以是任何序列长度的。可以将根据本发明的第二方面的核酸序列与感兴趣的基因组区域(靶标)连接以标记需要识别的物种和/或区分其群体内的标记物种的不同成员。因此,在本发明的一个方面的背景下,术语“索引序列”和“条形码”可以互换地使用。
如本文所用,术语“测序接头”是指已知序列的寡核苷酸,其与感兴趣的多核苷酸或多核苷酸链的连接或掺入使得能够产生准备扩增的所述感兴趣的多核苷酸或多核苷酸链的产物。
在本发明进一步的方面的一个实施方式中,所述核酸序列进一步包含至少一个切割位点。
如本文所用,术语“切割位点”是指易于由任何方式(包括但不限于能够切割单链或双链核酸序列的酶)被切割的核酸序列的靶区域。在本发明的背景下,所述“切割位点”可用于切割或以其他方式释放核酸序列的一部分。所述“切割位点”被切割剂识别,所述切割剂可以是天然的、合成的、未修饰的或修饰的。
在本发明的一个实施方式中,保护官能团选自在3’端上的间隔元件和在5’端上的双脱氧修饰碱基。在本发明的背景下,合适的非限制性间隔元件是三碳间隔子(c3间隔子)。
在本发明的另一个实施方式中,所述至少一个切割位点是包含回文区的限制性位点。
如本文所用,术语“限制性位点”是指被限制性内切酶(例如核酸内切酶)识别的位点。本领域技术人员熟悉限制性核酸内切酶及其限制性位点。限制性位点的非限制性实例包括bamhi、bsrl、notl、xmal、pspai、dpni、mbol、mnll、eco57i、ksp6321、dralll、ahall、smal、mlu1、hpal、apal、bcll、bsteii、taql、ecori、sacl、hindll、haell、drall、tsp509l、sau3ai、pacl。
在本发明的另一个实施方式中,所述核酸序列适合用于根据本发明的第一方面及其实施方式所述的方法。
在本发明的另一个实施方式中,所述核酸序列适合用于对从受试者获得的样品中的表观遗传状态进行谱分析。
如本文所用,术语“样品”是指生物样品。
如本文所用,术语“受试者”是指人或非人受试者。在后一种情况下,非人受试者不限于哺乳动物受试者。
根据本发明的方法可以在基因的识别,在受试者中的疾病状态的诊断和/或预后中涉及的因素,以及用于受试者中的疾病状态的诊断和/或预后和用于控制治疗性分子对染色质的作用的方法中找到不同应用。
在本发明的背景下,疾病状态可以指涉及核小体或核酸序列的任何修饰以及影响染色质结构、调节和功能的蛋白质的定位。如本文所用,表述“疾病状态”还涵盖细胞增殖的异常速率,其使得疾病的治疗需要调节细胞周期。增生性疾病的实例包括但不限于癌症。
根据本发明的方法可用于抗药性的体外诊断和/或预后,其中通过使用微流体系统对从受试者获得的细胞中的单细胞染色质状态进行谱分析,所述方法包括以下步骤:
a.提供至少第一类型的液滴,其中所述第一类型的液滴包含
i.生物要素,
ii.裂解缓冲液,和
iii.核酸酶,
b.在暂时使所述核酸酶失活的条件下收集所述第一类型的液滴,
c.孵育所述第一类型的液滴,从而重新激活所述核酸酶,
d.提供至少第二类型的液滴,其中所述第二类型的液滴包含核酸序列,
e.融合所述第一类型的液滴和所述第二类型的液滴,从而生成第三类型的液滴,
f.孵育所述第三类型的液滴,从而将所述核酸序列连接至一个或多个感兴趣的基因组区域,
g.对所述一个或多个感兴趣的基因组区域进行测序。
所述抗药性可能是新出现的抗药性和/或现存的抗药性。所述抗药性的出现可以归因于表观遗传异质性。
单细胞染色质谱分析表现为探查任何复杂生物系统内的染色质状态的异质性和动力学作用的独特工具:除癌症外,它还可被应用于其他疾病(特别是自身免疫疾病,传染性、代谢疾病)和健康系统,尤其是研究细胞分化和发展以及免疫监控。
根据本发明的方法可用于确定患者分层,其中使用微流体液滴中的单细胞表观遗传学谱分析将抗药性与不同染色质状态相关联是需要的。
在本发明的实施方式中,提供了一种用于处于疾病状态和/或怀疑处于疾病状态的受试者中的抗药性的诊断和/或预后的方法。
在本发明的实施方式中,提供了用于健康受试者中的抗药性的诊断和/或预后的方法。
根据本发明,可以在受试者接受治疗或疗法之前、期间或之后的时间点进行所述受试者中的抗药性的诊断和/或预后。所述诊断和/或预后也可以在任何其他时间点进行。所述治疗或疗法可能是使用化学治疗药物、化学药物或生物药物(例如抗体(及其衍生物或片段))的治疗或疗法,包括抗免疫检查点治疗,例如趋化因子,例如激素,例如细胞因子(及其衍生物),或例如由以下组成的细胞疗法:til(肿瘤浸润的t细胞)注射,cart细胞(嵌合相关抗原),carnk细胞,tcr疗法(以可溶性或细胞性疗法的形式),例如疫苗接种(癌症疫苗、病毒疫苗、诱导疫苗接种的树突状细胞疗法),例如溶瘤病毒,例如纳米粒子。
在本发明的实施方式中,提供了一种用于显示抗药性和/或怀疑具有抗药性的受试者的诊断和/或预后的方法。
所述受试者可以是处于疾病状态和/或被怀疑具有疾病状态的受试者或健康受试者。
如本文所用,术语“诊断”是指关于受试者是否可能显示或发生抗药性的确定。如本文所用的术语“诊断”是指本领域技术人员可以藉此估计和/或确定受试者患有抗药性(例如对治疗剂、化学治疗药物、化学药物或生物药物(例如抗体(及其衍生物或片段))的抗性的概率(“可能性”)的方法,包括抗免疫检查点治疗,例如趋化因子,例如激素,例如细胞因子(及其衍生物),或例如由以下组成的细胞疗法:til(肿瘤浸润的t细胞)注射,cart细胞(嵌合相关抗原),carnk细胞,tcr疗法(以可溶性或细胞性疗法的形式),例如疫苗接种(癌症疫苗、病毒疫苗、诱导疫苗接种的树突状细胞疗法),例如溶瘤病毒,例如纳米粒子)的方法。在本发明的情况下,“诊断”包括使用测定的结果,最优选scchip。
如本文所用,术语“预后”是指对于疾病(例如癌症,包括复发和转移性扩散、炎症、感染性疾病、自身免疫疾病、代谢疾病、遗传和非遗传疾病)的可归因于抗药性的死亡或发展的可能性的预测。
根据本发明的方法可使用源自身体样品的单细胞。
在本发明的实施方式中,所述身体样品是流体和/或固体。如本文所用,所述身体样品可以来自组织、血液、血清、血浆、唾液、粪便、尿液、乳房、肺、结肠、肠、脑、结肠、肾脏或任何其他身体样品。
根据本发明,所述一个或多个感兴趣的基因组区域是修饰的基因组区域。所述修饰的基因组区域包含核酸序列和/或与核酸序列相关联的蛋白质复合物。所述修饰的基因组区域包括翻译后修饰,所述翻译后修饰选自乙酰化、酰胺化、脱酰胺化、羧化、二硫键、甲酰化、糖基化、羟基化、甲基化、肉豆蔻酰基化、亚硝基化、琥珀酰化(assuccinylation)、丁酰化、磷酸化、异戊烯化、核糖基化、硫酸化、sumo化、泛素化及其衍生物。
在优选的实施方式中,用化学治疗药物、化学药物或生物药物(例如抗体(及其衍生物或片段),包括抗免疫检查点治疗,例如趋化因子,例如激素,例如细胞因子(及其衍生物)或例如由以下组成的细胞疗法:til(肿瘤浸润的t细胞)注射,cart细胞(嵌合相关抗原),carnk细胞,tcr疗法(以可溶性或细胞性疗法的形式),例如疫苗接种(癌症疫苗、病毒疫苗、诱导疫苗接种的树突状细胞疗法),例如溶瘤病毒,例如纳米粒子)处理所述细胞。在更优选的实施方式中,用化学治疗药物处理所述细胞。
在本发明的一个实施方式中,所述细胞是用他莫昔芬(其靶向雌激素受体)和卡培他滨(经典的抑制胸苷一磷酸合成的化学疗法)来处理的。
在本发明的一个实施方式中,所述细胞未经处理。
如本文所用,“疾病状态”是指疾病,例如癌症或传染性疾病、自身免疫疾病、代谢疾病、炎症疾病、遗传和非遗传疾病。
在本发明的一个实施方式中,受试者的疾病状态包括癌症、感染性疾病、自身免疫疾病、炎症疾病、代谢疾病、遗传疾病、非遗传疾病。
在本发明的一个实施方式中,疾病状态包括从不可检测的至第四期的任何阶段的癌症。在本发明的一个实施方式中,癌症包括任何类型的癌症,例如实体和/或液体癌症。
在本发明的优选实施方式中,受试者的疾病状态是乳腺癌。
根据本发明,受试者可能是雄性或雌性受试者。在本发明的优选实施方式中,所述受试者是雌性受试者。
在根据本发明的进一步实施方式中,所述单细胞染色质状态已经丢失了促进抗药性的基因的染色质标记物。所述染色质标记物包括不同组蛋白修饰h3k4me3、h3k27ac和h3k27me3。预期h3k4me3标记物允许基因不被永久沉默和使其在需要时被激活。预期h3k27me3标记物可使所述基因沉默。预期在基因增强子处的h3k27ac标记物促进基因激活。
在一个实施方式中,所述单细胞染色质状态已经丢失了可能导致抗药性的基因的染色质标记。染色质标记包括组蛋白修饰h3k4me3和h3k27me3。
如本文所用,“染色质标记物”、“基因的标记物”或“标记物”是指有助于在不同染色质状态内定义表观基因组特征的dna和组蛋白修饰,其高度指示细胞类型和组织身份。可以利用这些标记物的全基因组谱分析来了解基因组调控的全局景观,然后例如区分在正常和疾病细胞状态的背景下的表观基因组差异。技术人员知道如例如在(consortiumepigenomics2015,nature518(7539):317-329)或其他研究中公开的若干染色质标记物。
在一个实施方式中,单细胞染色质状态已获得标记物,其中所述标记物具有去沉默作用。
如本文所用,术语“癌症”和“肿瘤”可互换地使用且涉及恶性肿瘤形成。恶性肿瘤形成的实例包括实体瘤和血液肿瘤。实体瘤由乳腺、膀胱、骨骼、脑、中枢和周围神经系统、结肠、内分泌腺(例如甲状腺和肾上腺皮质)、食道、子宫内膜、生殖细胞、头颈、肾脏、肝、肺、喉和下咽、间皮瘤、卵巢、胰腺、前列腺、直肠、肾脏、小肠、软组织、睾丸、胃、皮肤、输尿管、阴道和外阴的肿瘤示例。恶性肿瘤包括由视网膜母细胞瘤和威尔姆氏瘤(wilmstumor)示例的遗传癌症。此外,恶性肿瘤包括所述器官中的原发性肿瘤和远处器官中的相应的继发性肿瘤(“肿瘤转移”)。血液肿瘤由白血病和淋巴瘤的侵袭性和惰性形式示例,即非霍奇金斯病、慢性和急性髓性白血病(cml/aml)、急性淋巴细胞白血病(all)、霍奇金斯病、多发性骨髓瘤和t细胞淋巴瘤。还包括骨髓增生异常综合症、浆细胞肿瘤形成、副肿瘤综合症、原发部位未知的癌症以及与艾滋病相关的恶性肿瘤。
为了确定癌症已发展的程度,通常在诊断时的生长和扩散方面以第一至第四期对癌症进行标记。在第一期中,癌症局限于身体的一个部位且可通过手术切除。在第二和第三期中,癌症是局部晚期的且可通过化学疗法、放射或手术来治疗。在第四期中,癌症已经转移或扩散至其他器官且可通过化学疗法、放射或手术来治疗。(第五期仅用于受威尔姆氏肿瘤影响的患者,其中两个肾脏均受影响。)
如本文所用,“代谢疾病”包括但不限于代谢综合症x、先天性代谢错误、线粒体疾病、磷代谢紊乱、卟啉症、蛋白原代谢缺乏症、代谢性皮肤病、消耗性综合症、水-电解质失衡、代谢性大脑疾病、钙代谢紊乱、dna修复-缺乏紊乱、铁代谢紊乱、脂质代谢紊乱、吸收不良综合征。
如本文所用,“自身免疫疾病”包括但不限于多发性硬化症,淀粉样变性病,强直性脊柱炎,抗gbm/抗tbm肾炎,抗磷脂综合征,自身免疫血管性水肿,自身免疫自主神经异常,自身免疫脑脊髓炎,自身免疫肝炎,结节性多发性动脉硬化,i、ii、iii型多腺体综合征,风湿性多肌痛,多发性肌炎,发作性睡病,坏疽性脓皮病,雷诺现象,间质性膀胱炎(ic),青少年关节炎,青少年糖尿病(1型糖尿病),青少年肌炎(jm),反应性关节炎,新生儿狼疮,视神经脊髓炎,腹腔疾病,恰加斯病(chagasdisease),原发性胆汁性肝硬化,原发性硬化性胆管炎,孕激素性皮炎,牛皮癣,牛皮癣性关节炎,慢性炎性脱髓鞘性多发性神经病(cidp),横贯性脊髓炎,1型糖尿病,溃疡性结肠炎(uc),慢性复发性多病灶性骨髓炎(crmo),churg-strauss综合征(css)或嗜酸性肉芽肿病(egpa),嗜中性白血球减少症,眼瘢痕性天疱疮,视神经炎,回文型风湿症。
如本文所用,“遗传疾病”包括但不限于重症联合免疫缺陷(scid)、镰状细胞病、皮肤癌、威尔逊病、特纳综合症(turnersyndrome)、脊髓性肌萎缩症、tay-sachs、地中海贫血、三甲基尿失禁、强直性肌营养不良症、神经纤维瘤病、努南(noonan)综合征、痛性肥胖病(dercumdisease)、唐氏综合症、duane综合症、杜氏肌营养不良症(duchennemusculardystrophy)、莱登第五因子血友病(factorvleidenthrombophilia)、自闭症、常染色体显性多囊肾病、乳腺癌、腓骨肌萎缩症(charcot-marie-tooth)。
如本文所用,“炎症疾病”包括但不限于过敏、哮喘、自身免疫疾病、腹腔疾病、肾小球肾炎、肝炎、炎性肠病。
实施例
单细胞chip-seq程序的微流体工作流程
在图1中描绘了根据本发明的微流体方法的一般方案。(a)在45pl液滴中用细胞裂解和染色质片段化所需的试剂将细胞区室化。平行地,将带有dna条形码的水凝胶珠粒与连接试剂和使微球菌核酸酶失活的egta一起包封于100p1液滴中。在融合设备中重新注入两种乳液,将包含条形码的液滴(100p1)和包含核小体的液滴(45p1)非对称地配对,并通过电场触发的电聚集来融合。用激光束对融合液滴进行一对一扫描以实时分析每个液滴的组成。(b)收集融合液滴的乳液,并在芯片外孵育,以用于在液滴中进行核小体条形码化。通过光切割从珠粒释放条形码并将其连接至核小体。合并液滴的内容物,进行免疫沉淀,并对富集的dna进行测序。与条形码相关的读数的反卷积将所有序列归属于其原始细胞以重建单细胞染色质谱分析。
同步和暂停液滴中的染色质片段化
在45pl液滴中用包含裂解缓冲液和微球菌核酸酶的消化共混物将细胞区室化(参见图1)。在完全裂解之后,染色质被释放至液滴中,其可被微球菌核酸酶切割。本节介绍了液滴中微球菌核酸酶活性的典型校准以优先地产生具有核小体的尺寸的片段。然而,由于细胞是以不同时间尺度单独处理,因此在液滴中进行酶促测定可能具有挑战性。因此,微调酶活性以避免液滴和单细胞之间的染色质消化差异是必要的。
液滴中的细胞的区室化
每液滴细胞数量遵循泊松分布,其描述了发现每液滴x个细胞的平均数量λ的概率(howardshapiro,practicalflowcytometry,第4版,wiley-liss,2003)。在单细胞chip-seq实验中,调整细胞密度以在45pl液滴中包封λ=0.1个细胞,从而导致90.5%的空液滴,9%包含一个单细胞的液滴,0.5%包含两个细胞的液滴和0.015%包含多于两个细胞的液滴。通过用钙黄绿素am(钙黄绿素的非荧光衍生物)预先标记细胞来进行对液滴中的细胞的区室化的实时监控。在进入细胞之后,乙酰甲氧基(am)被细胞内酯酶切割并释放出强烈的绿色荧光(激发/发射:495/515nm)。当液滴在检测点处穿过激光束时采集荧光,从而允许对包封的细胞的数量进行计数。
液滴收集
将液滴收集在冰上的收集管中直至包封结束(10分钟至20分钟,具体取决于起始细胞的数量)。包封后,将液滴在37℃下孵育用于微球菌核酸酶消化。
液滴中的微球菌核酸酶校准
在包封结束时,将液滴在芯片外孵育以用于单细胞染色质片段化。在液滴中裂解细胞,使它们的核dna可用于微球菌核酸酶。该消化的动力学对于优先地产生保留在液滴中的单核小体特别重要。理想的孵育时间定义为将100%的核dna片段化为单核小体所必需的时间。通过进行时程研究,精确校准每个样品的消化条件(包括裂解缓冲液组成、微球菌核酸酶浓度和孵育时间)。如下进行校准:生成包含细胞、缓冲液和微球菌核酸酶的45pl液滴,将其收集在收集管中,并将其在37℃下放置不同的孵育时间。在每个时间点,一定分数的液滴被破裂,并通过添加egta使微球菌核酸酶立即失活(参见图3)。然后纯化dna片段并通过电泳对其进行分析。孵育时间的选择是在具有最高比例的单核小体但同时防止核小体dna被过度消化之间的平衡。确实,假设是从核小体突出的dna应该足够长以使得在该程序的后续步骤中能够进行条形码的有效连接。
控制液滴中的微球菌核酸酶活性
通过在细胞包封之后在冰上收集液滴来控制液滴中的微球菌核酸酶活性(参见图2)。确实,图2中的时间点t=0分钟(其对应于液滴生产结束时但就在孵育之前所采集的一定分数的液滴)表明核dna尚未被微球菌核酸酶消化。该证据确认了染色质消化不在液滴产生时发生,而是在37℃的孵育下立即开始(参见图2)。
在孵育之后以及在融合设备中重新注入时将液滴置于冰上可“暂停”微球菌核酸酶活性并限制染色质消化中的细胞间变化。为了这个目的,在12分钟的微球菌核酸酶孵育之后取两个液滴部分:一个部分被立即处理以控制消化,而第二部分被预先存于冰上1小时,然后进行类似处理。如预期的,图2上的时间点t=12分钟和t=12分钟+1小时冰确认了微球菌核酸酶在冰上存储的部分中不再具有活性(相比于t=20分钟的时间点)。因此,将液滴存储在冰上“暂停”了微球菌核酸酶活性,从而防止液滴之间的染色质消化再次发生变化。
dna条形码化策略
通过链霉亲和素-生物素连接和可光切割部分将dna条形码接合至水凝胶珠粒,使得其在暴露于紫外线时能够从珠粒释放(klein等,2015,cell161(5):1187-1201)。条形码的合成包括将微珠分布于包含连接试剂和20bp寡核苷酸的96种组合(后称为索引1)的微孔板中。将索引1连接至珠粒并将其合并,之后再次分布于包含20bp寡核苷酸的96种新组合(后称为索引2)的第二微孔板中。通过重复此拆分-合并方法3次,轻松生成963种可能的条形码组合的文库(即884,736种组合)。
条形码化水凝胶珠粒的质量控制
条形码化珠粒是scchip-seq技术的核心试剂之一,其质量已得到系统地控制以确保细胞间变化是源自其组蛋白修饰模式中的真正生物差异,而不是技术伪像。
从珠粒释放的dna条码的tapestation谱分析显示,>75%是全长(146bp处的较大峰),以及未能完整的中间体的存在(图5)。平均而言,全长条形码的数量被估计为每条形码化水凝胶珠粒5×107个拷贝。
为了验证条形码从水凝胶珠粒的释放,将dna探针与条形码杂交至珠粒上。然后将后者包封于100p1液滴中,并如scchip-seq实验中那样芯片外收集。如eyer(eyer等,2017,naturebiotechnology35(10):977-982)报道的,将部分液滴作为单一列队(singlefile)重新注入至测微室中,并通过辐射荧光显微镜对微珠成像,同时荧光条形码仍然结合在珠粒上。如预期的,荧光定位在珠粒上(参见图5)。将第二部分的包含珠粒的液滴暴露于紫外线以引发条形码释放。如上所述,在光切割之后的包含珠粒的液滴的辐射荧光显微显示了液滴中荧光的均匀分布,其表明完全的条形码释放(参见图5)。最后,对每一批新的条形码化珠粒进行单珠测序。通过在384孔板中进行有限稀释来分离珠粒。通过成像仅选择包含一个珠粒的孔以用于条形码的扩增和测序。测序数据的分析显示了16个珠粒的前两个最丰富的条形码的分数。识别出每珠粒数十万种不同的条形码,但平均而言,最丰富的条形码占测序读数的97.7%。第二丰富的条形码平均占读数的少至0.17%,这表明所有其他识别出的条形码是可忽略不计的(参见图5)。
条形码设计
条形码通过链霉亲和素-生物素连接与珠粒结合,其进而通过可光切割实体与寡核苷酸的5’端分开。后者包含可光切割基团和使空间相互作用最小化的烷基间隔基(整个实体被称为pc-接头,参见图4)。第一生物素化的和pc接头寡核苷酸是所有条形码共有的,并包含t7启动子序列和illumina测序接头(sbs12序列)。t7启动子序列充当t7rna聚合酶的识别位点以在体外转录反应(ivt)中启动免疫沉淀后富集的条形码化核小体的线性扩增。在单细胞rna-seq方案中的逆转录后的cdna的无偏倚、灵敏和可再现扩增中广泛采用了这种扩增策略(hashimshony等,2012,cellreports2(3):666-673)。在第二步中,illumina测序接头充当pcr手柄以完成测序文库的制备。并且,此接头作为启动读数#2和对条形码序列的读取的引物对于样品的下一代测序而言是必需的。用该第一共有寡核苷酸接合的珠粒然后通过连续连接3个索引来用于条形码合成。不幸的是,对第一单细胞chip-seq数据集的分析显示,只有很少的读数(~38%)具有完整且正确的条形码结构。
在图3中描绘了优化的条形码结构,其允许消化条形码多联体以及减少非全长条形码的连接。条形码用pac1限制性位点的一半框起来,其仅在形成多联体的情况下才被重建。那些在免疫沉淀之后但在线性扩增之前被消化以清理文库。通过引入3’c3间隔子修饰条形码光切割侧。该修饰在3’碱基的3’-羟基处引入间隔臂并阻断连接。随着间隔基的添加,连接的方向被强制为是从条形码的3’-端至核小体。非全长条形码用包含3’c3间隔子和5’反向二脱氧-t碱基的“嵌段”寡核苷酸序列来完整。再次地,两种修饰均目的在于限制不需要的连接事件。
条形码化珠粒在液滴中的包封
可以通过泊松分布来估计将离散物体(例如水凝胶珠粒)加载至液滴中。以与细胞的包封相同的方式,在液滴在检测点处穿过激光束时,实时监控珠粒的加载。在单细胞chip-seq实验中,其通常实现65%至75%的包含条形码化水凝胶珠的液滴。
包含核小体的液滴与包含条形码的液滴融合
将细胞和dna条形码分别包封以防止条形码被微球菌核酸酶消化。为了在单细胞水平上对染色质进行索引化,必须在第二步中将dna条码递送至包含核小体的液滴中。这是通过使用触发电场在专用微流体设备中主动融合两个液滴群体来实现的。
来自“细胞乳液”的液滴和来自“条形码乳液”的液滴作为单一列队重新注入微流体融合设备中。实现适当的电聚集需要将来自两种乳液的液滴一对一配对。水动力使更快的且较小的45pl液滴(“细胞乳液”)能够赶上100pl液滴(“条形码乳液”)并与其接触,因为接触对于两个液滴融合是必须的(mazutis等,2009,labonachip9(18):2665)。与液滴产生相似,在融合液滴在检测点处穿过激光束时获得其荧光强度(参见图1)。
从单细胞chip-seg谱分析重建细胞类型特异性染色质状态
如图7所示,将人t淋巴细胞和人b淋巴细胞分别包封,并用两组不同的条形码来索引化。在液滴中对核小体进行条码化之后,将来自两种细胞类型的索引化染色质合并,进行染色质免疫沉淀并对文库进行测序
通过合并索引化染色质,避免了引入与测序文库的免疫沉淀或制备有关的偏倚(批次效应)。每个测序读数会携带双重信息:(1)单细胞条形码序列,其将读数分配给其起源细胞;(2)“细胞类型特异性序列”,其将读数分配给一种细胞类型(b或t淋巴细胞)。
为了确认条形码对于单个细胞是独特的,已经用小鼠和人细胞系的混合物进行了实验,其显示97%的条形码被毫无疑义地分配给单个物种,其与已占用的包含单个细胞的液滴的百分比(95%)一致,如图13所示。
已验证scchip-seq程序从h3k4me3和h3k27me3修饰的单细胞分布重述细胞身份的效率和准确率。使用两组独立的条形码化接头分别处理人ramos(b细胞)和jurkat(t细胞)(如图1a-b所示),并在液滴中连接接头之后,合并条形码化核小体并对其进行免疫沉淀。对于h3k4me3和h3k27me3组蛋白标记物,分别实现了每细胞1,630和1,633个独特读数的平均覆盖率,和跨技术和生物重复实验的高相关性(图14a-c,分别为r=0.96和0.98,p<10-15)。
对于单细胞染色质谱分析实验两者,通过一致性聚类识别了对应于每个细胞系的两个稳定聚类(图15a和图16a),将细胞身份与对h3k4me3和h3k27me3谱分析分别为超过99.7%和99.5%的特异性相匹配。聚集的单细胞谱分析以高准确率重述了成批chip-seq谱分析(图15b-c,对于h3k4me3和h3k27me3,分别为r=0.93和0.97,p<10-15,图14f)。通过差异性分析识别了对ramos和jurkat细胞具有特异性的许可性和抑制性染色质特征(图16b)。聚焦于h3k4me3,其在转录起始位点附近积累,我们识别了多组协调的谱系特异性基因,如在对每个细胞系具有特异性的染色质特征中富集的(图16c)。这些结果确认,scchip-seq程序是在单细胞水平上检测染色质景观,根据其染色质状态以高准确率对单细胞进行分类,并识别细胞群体之间的区别性染色质特征的稳健方法。
单细胞条形码和细胞类型特异性序列的反卷积
通过首先搜索在条形码的20聚索引之间发现的恒定4bp接头来从读数#2中提取条形码,从而允许每个接头中最多1个错配。如果识别了正确的接头,则提取三个散布的20聚索引并将其连接在一起以形成60bp的非冗余条形码序列。使用灵敏读数映射器cushaw3,用3组96索引(963)的所有884,736个组合的文库来映射条形码序列。每组索引都是错误修正的,因为将一个索引转换为另一个索引花费超过为3的编辑距离。因此,将在整个条形码中的总错配阈值设置为3,每索引为2或更少,以避免将序列错误分配给错误的条形码id。在第二个较慢的步骤中,将不可以映射至cushaw3索引文库的序列拆分成其个体索引,将每个索引与96个可能索引的集进行比较,从而允许每个个体索引中最多2个错配。未通过这两个步骤分配给条形码id的任何序列都被丢弃。
单细胞chip-seq数据分析
使用参比基因组hg38(genbank集合登记:gca_000001405.15),用python(v2.7.12)和r(v3.3.3)对测序数据进行了分析。
多路解编细胞条形码—比对、过滤和标准化
通过仅保留具有不超过一个可报告的比对和2个错配的读数,使用bowtie(v.2.2)将读数#1与小鼠mm10和人hg38参比基因组进行比对。
原始读数是根据双峰分布而分布的,较低的峰最可能对应于具有条形码化珠粒但没有细胞的液滴(rotem,a等,natbiotechnol;2015,33:1165-1172),以及右边的峰对应于具有珠粒和细胞的液滴(图6a);从而设置读数计数截止点以定义与细胞相关联的条形码。为了后续分析,本发明人将条形码的独特的(pcr重复去除之后的)读数计数保持在该临界点之上。为了去除pcr重复,对于每个条形码(即细胞),将落在相同150bp窗口中的所有读数堆叠在一起为一个,作为可能源自pcr重复或同一核小体的读数。本发明人从这些去重复的读数中生成了覆盖矩阵和度量,其在本文中被称为“独特读数”。
对于每个细胞,读数被归入(binned)h3k27me3的不重叠的50kb(已知在整个宽的基因组区域内积累)和h3k4me3的5kb基因组分组(bin)(已知在转录起始位点附近的窄峰内积累)中,跨越基因组以产生具有n个条形码和m个基因组区间的nxm覆盖矩阵。本发明人合并了来自以下样品的四个分析中每一个的覆盖矩阵:(i)ramos和jurkat,(ii)来自hbcx95和hbcx-95-capar的小鼠细胞,(iii)来自hbcx95和hbcx95-capar的人细胞,和(iv)来自hbcx-22和hbcx-22-tamr的人细胞。
本发明人首先去除了具有在较高百分位数内的独特映射读数的总数(被视为离群值)的细胞,并滤出了未代表所有细胞的至少1%的基因组区域。仅当细胞具有每细胞至少1,600个独特读数时,本发明人才可通过pca分析来对细胞进行分组而不依赖于覆盖率。对于所有后续分析,本发明人排除了具有较低覆盖率的细胞。然后通过将计数除以每个细胞的读数总数并乘以所有细胞的平均读数数来对覆盖矩阵进行标准化。
单细胞chip-seq图谱的无监控聚类
通过主成分分析减少了标准化的矩阵(n=50,用于进一步分析的第一成分)。为了提高聚类方法的稳定性,本发明人进一步将分析限于至少1%的细胞显示出高于阈值t的皮尔森成对相关性得分(pearson’spairwisecorrelation)的细胞。阈值t定义为随机数据集的皮尔森成对相关性得分的上百分位数。
本发明人使用一致性聚类,bioconductorconsensusclusterplus软件包(wilkerson,m.d.&hayes,d.n.2010.bioinformatics26,1572-1573),检查了簇的稳定性并计算了每个细胞的项目一致性得分。本发明人基于层次聚类的1,000个重采样迭代(80%的细胞),以皮尔森的相异性作为距离度量并将ward方法用于连接分析,建立了k(k=2、3……)个簇的数据集的一致性分区。选择了最佳的簇数(k)以最大化簇内相关性得分。聚类结果通过t-sne图可视化(vandermaaten,l.&hinton,g.2008.jmachlearnres9,2579-2605)。为了可视化亚群体的染色质图谱,本发明人聚集了每个簇内单细胞的读数,并使用r软件包sushi创建了富集图谱(phanstiel,d.h.等,2014.bioinformatics.30,2808-2810)。
单细胞chip-seg图谱的差异分析
为了识别给定簇的单细胞之间的差异富集区域,本发明人进行了非参数威尔科克森秩和检验,比较了来自一个簇的单个细胞的标准化计数与来自所有其他细胞的标准化计数。本发明人检验了零假设,即来自两个比较组的标准化计数的分布具有相同的中位数,置信区间为0.95。本发明人将分析限于选择用于上文所述的无监控分析的窗口。
使用benjamini-hochberg方法对p值进行多次测试校正(benjamini,y.&hochberg,y.1995.jrstatsoc57,289-300)。如果经调节p值、“q值”小于0.01并且绝对log2倍数变化大于1,则h3k27me3或h3k4me3的基因组区域被视为“富集的”或“耗竭的”。
scrna-seq比较。
对于h3k27me3scchip-seq分析,本发明人使用了来自总chip-seq数据集的峰注释来进一步注释50kbp窗口和对应的基因:对于每个窗口,本发明人保留了其以用于后续分析(基因注释和scrna-seq比较)的基因;对于具有在任何情况下与峰重叠的转录起始位点(tss)的基因,使用bedtools(v2.17)50和人转录组gencode_hg38_v26(限于蛋白质编码)、反义和incrna基因的参照注释。
成批chip-seq
如先前在vallot,c.等,2015.cellstemcell16,533-546中描述的,对来自上文使用抗h3k27me3抗体(cellsignalingtechnology,#9733)而从hbcx-22、hbcx-22-tamr、hbcx-95和hbcx-95-capar获得的细胞悬液的106个细胞进行chip实验。根据制造商的说明,使用ovation超低文库系统v2(nugene),使用2ng的免疫沉淀的和输入的dna来制备测序文库。在illuminahiseq2500上以快速运行模式(rapidrunmode)se50对成批chip-seq文库进行测序。
总chip-seq数据分析
使用bowtie(v.2.2)将读数与小鼠mm10和人hg38参比基因组进行比对,并使用工具bamcmp来将人与小鼠序列分离。如前文所说明的那样进行后续分析。仅保留独特映射读数以用于分析。此外,使用picard工具(https://broadinstitute.github.io/picard/)删除了pcr重复。数据被合并至5kb(h3k4me3)或50kb(h3k27me3)的连续的基因组窗口中。对于每个窗口,计算log2rpm作为每百万映射读数的记录的读数log数。
单细胞rna-seq
根据制造商的说明,将来自每种细胞悬液hbcx-22、hbcx-22-tamr、hbcx-95和hbcx-95-capar的约3,000个细胞装载至chromium单细胞控制仪(10xgenomics)上。根据制造商的说明来制备样品和文库。使用配对端26bp-98bp测序来在illuminahiseq2500上以快速运行模式对文库进行测序。
单细胞rna-seq数据分析
使用cellrangersinglecellsoftwaresuite(v1.3.1)处理单细胞测序图谱,以执行质量控制、样品多路解编、条形码处理和单细胞3’基因计数(http://software.10xgenomics.com/single-cell/overview/welcome),其使用具有默认参数的ucsc小鼠(mm10)和人(hgl9)转录组和基因组。对细胞进行分析,在所述细胞中,2,728个(1,564个人细胞和1,191个小鼠细胞)细胞对hbcx-22的平均覆盖率为30,166个读数/细胞,1,746个细胞(753个人细胞和1,013个小鼠细胞)对hbcx-22-tamr的平均覆盖为41,166个读数/细胞,1,184个细胞(545个人细胞和647个小鼠细胞)对hbcx-95的平均覆盖率为160,583个读数/细胞,2,087个细胞(861个人细胞和1,242个小鼠细胞)对hbcx-95-capar的平均覆盖率为38,345个读数/细胞。使用自定义r脚本在r(v3.3.3)中进行了进一步的分析。筛选出线粒体umi计数超过10%的任何细胞。本发明人只保留了umi总数低于100,000的细胞以及检测到的基因低于6,000且超过1,000的细胞。然后,本发明人仅将具有至少1个转录物的基因保留在至少2个细胞中。使用rpackagescater,对scrnaseq计数矩阵进行相对于覆盖率的标准化并通过“相对log表达”方法来对其进行转化(mccarthy,d.j等,bioinformatics,2017,33:1179-1186)。使用来自r软件包ccremover的注释(barron,m.等,scirep,2016,6:33892),本发明人从后续聚类分析中去除了与细胞周期相关的基因以根据细胞身份而非与细胞周期相关的现象对细胞进行分组。然后在n=50的第一主成分(pca)上进行至t-sne的barnes-hut逼近以可视化二维空间中的细胞。与上述scchip-seq分析一样,使用一致性聚类确定簇。本发明人使用edgerglm统计模型识别了簇之间差异表达的基因(robinson,m.d.等,bioinformatics,2010,26:139-140)。对于基质小鼠细胞,根据标志基因的差异表达识别了簇。
总肿瘤细胞的拷贝数分布
r软件包hmmcopy用于校正未经治疗的与抗性异种移植模型中的拷贝数变化。来自总输入chip-seq样品的读数被合并至横跨整个基因组的0.5mb非重叠区域中。去除平均偏差大于n=2标准差的区域以进行分析。
本文之后显示了一个实例,其中本发明的方法通过通过使用微流体系统对从受试者获得的细胞中的单细胞染色质状态进行谱分析而用于抗药性的诊断和/或预后。
本文显示了scchip-seq实验如何可在对化学疗法的获得性抗性的三阴性乳腺肿瘤模型中预测染色质标记的异质性。
如本文所用,“三阴性肿瘤”是指以er-,pr-和her2-为特征的乳腺癌。
对于根据本发明所述的方法,在对来自单个患者的异种移植衍生物进行两个周期的治疗之后(最初对卡培他滨起反应)获得了对卡培他滨具有抗性的肿瘤hbcx-95-capar(图17a)。针对敏感和抗性异种移植物hbcx-95和hbcx-95-capar,以单细胞分辨率对h3k27me3的谱分析进行了分析(图17b),并且还进行了scrna-seq以评估同一细胞悬液中的转录异质性(图18a)。累积的单细胞染色质谱分析与成批chip-seq谱分析相匹配(图17c-d,r=0.89,p<10-15)。
研究了基质细胞(n=1766个小鼠细胞,平均每个细胞3535个独立读数的平均覆盖率)内染色质图谱的多样性。一致性聚类方法(图19)表明,根据h3k27me3分析(而不论患者来源的异种移植物(pdx)是否对治疗敏感或对其具有抗性),基质细胞稳定地分为三个基于染色质的群体chrom_cl、c2和c3(图18b),其反对潜在的批次效应。通过比较细胞组之间的染色质特征(图20),确定了对chrom_c2和c3群体具有特定h3k27me3富集(n分别为1,581和n=1,282,q值<0.01和|log2fc|>1)而对簇chrom_c1(n=122)程度较小的位点。
同时,scrna-seq分析显示了四个基质细胞群体(图18c和图21):两组成纤维细胞来源的细胞(具有特定标记col12al和efempl),内皮细胞(pecam-1)和巨噬细胞(ms4a7)。为了进一步比较从两种方法推断出的群体身份,特别关注的是转录起始位点位于chrom_c1,c2和c3特有的染色质特征1kb内的基因。在来自chrom_c2的细胞中明确不含h3k27me3的位点,即仅允许在chrom_c2中转录的位点在上皮到间质转化中涉及的基因(q值=2.8x10-3)(例如col4al(图20b))中显著富集,或在顶端连接处涉及的基因(q值=9.0×10-2)(例如ptk2(图18d))中显著富集;这两个特征都是成纤维细胞表达程序的特征。类似地,发现缺乏对chrom_c3具有特异性的h3k27me3的基因座,该基因座富包含来自免疫表达程序(q-值=5.2×10-2,图20a)的基因,例如lrmp(图18e)。scrna-seq数据进一步证实了这些标记在pdx样品中的表达模式(图18d-e的右图)。未能识别出与chrom_c1的几个染色质标记特征相关的相关基因,这表明用本发明的scchip-seq程序捕获这些细胞的效率较低,或者这组细胞与chrom_c2和c3都具有染色质特征。实际上,来自该簇的细胞中有一半与ptk2的免疫样细胞h3k27me3富集共享(图18d)。总而言之,根据本发明的scchip-seq方法揭示了小鼠基质细胞内存在三个h3k27me3染色质景观,其中两个与用scrna-seq识别的转录组特征(成纤维细胞和免疫样特征)匹配。
接下来,研究了来自同一对三阴性乳腺肿瘤样品的肿瘤细胞之间染色质分布的异质性(n=来自hbcx-95和hbcx-95-capar的4,331个细胞,每个细胞平均覆盖5,161个独特读数)。从分析中去除了如从成批dna谱分析中识别以集中在染色质变化上的受拷贝数变化影响的基因座(图22a)。基于染色质和转录组谱分析两者,细胞主要根据其敏感或抗性肿瘤起源而聚类(图23a-c和图22b-c)。虽然敏感细胞的染色质分布在很大程度上是同质的,但抗性群体内的染色质状态却很明显(图23a),这表明出现了具有明显染色质特征的抗性细胞的异质群体。但是,一致性聚类还显示,来自未经治疗的肿瘤的细胞中有3%(n=484个中的13个)稳固地与抗性细胞归为一类(图23d,一致性得分超过0.9),这表明它们具有共同的染色质特征。与chrom_c2相对应的抗性样细胞和抗性细胞与来自chrom_c1的敏感细胞相比,在h3k27me3中显示出大量的基因座缺失(图23e-f,n=569个具有耗竭的h3k27me3的基因座对114个具有富集的h3k27me3的基因座,q值<0.01,且|log2fc|>1,其中30%与转录起始位点重叠)。chrom_c2细胞中特别缺乏h3k27me3的基因座富含polycomb复合物的基因靶标(图22d),这表明预期的ezh2靶标的脱甲基化。由于缺少转录或由于scrna-seq程序的敏感性不足,只能检测到这些基因座的5%以内的转录。在这些基因座中,六个基因根据scrna-seq显著失活,并且全部根据其h3k27me3染色质状态而失控(图23f,图22e)。有趣的是,识别了igf2bp3(已知可促进对化学疗法的抗性的基因(图23g))和具有诱导对化学疗法的抗性的上皮至间充质转变的标记的区域(col4a2,hoxd簇,图23h-i)。
此外,还对一对管腔er+乳腺pdx进行了谱分析:hbcx-22对他莫昔芬和hbcx-22-tamr有反应,其是之前以对他莫昔芬的获得性抗性为特征的肿瘤衍生物。为了获得每个细胞10228个独特读数的高平均覆盖率,限制了包封细胞数(n=822个肿瘤细胞,图25a-b)。肿瘤细胞显示出与它们的起源肿瘤相关的两个主要染色质特征。但是,16%(255个中的n=41)的敏感肿瘤内的细胞与所有抗性细胞均具有染色质特征(图24a-c和图25c)。已经在敏感肿瘤的罕见细胞中发现了具有抗性细胞特征的染色质特征,并且可已被他莫昔芬治疗所选择。
染色质特征的差异分析显示,与敏感样细胞相比,抗性样细胞和抗性细胞(chrom_c2)主要丢失了h3k27me3标记(图25d,n=356个具有耗竭的h3k27me3的基因座对137个具有富集的h3k27me3的基因座)。特别缺乏来自chrom_c2的细胞中的h3k27me3的基因座在polycomb复合物的基因靶标中富集,并且具有乳腺上皮的基底样签名的特征(图25e)。使用scrna-seq,只能在2%的差异富集窗口中检测到转录和3个基因的显著差异表达,其均显示出在抗性细胞的馏分中的与h3k27me3富集对应的转录激活:egfr(涉及对他莫昔芬的抗性)、igfbp3和alcam(图24d、h-i和25g)。
相同细胞悬液的平行scrna-seq分析显示抗性和敏感肿瘤中的若干簇(图24e-f,图25f)。尽管没有来自敏感肿瘤的细胞与抗性细胞聚类,本文显示来自rna_c6簇的细胞(源于敏感肿瘤)(对应于1,275个细胞中的211个细胞,占17%)显示出抗性肿瘤细胞特有的途径的激活,其中包括基底样基因签名和上皮到间充质转化的签名(图24g)。这些观察结果独立地证实,在转录组或染色质水平下在来自敏感肿瘤的细胞中发现了抗性细胞共有的非遗传特征。两种单细胞测量均指向基底样基因签名的激活,但是通过不同的基因集。
以高达平均每细胞10,000个基因座的高覆盖率在单细胞水平上对组蛋白修饰进行谱分析有助于揭示肿瘤样品内相对罕见的染色质状态的存在。该单细胞染色质谱分析表现为探查任何复杂生物系统内染色质的异质性和动力学的作用的独特工具:除癌症外,其还可被应用于其他疾病和健康系统,尤其是研究细胞分化和发展用于患者分层。
根据本发明的方法可以用于揭示具有抗性癌细胞特有的染色质特征的罕见的细胞在治疗之前存在,并可通过癌症疗法对其进行选择。如前所述,未经治疗的细胞中染色质状态的自发异质性可能是获得抗性的关键分子成分,而与癌症治疗的作用机制无关:本文中靶向雌激素受体的他莫昔芬和经典的卡培他滨化学疗法可抑制胸苷一磷酸的合成。
根据本发明所述的方法揭示了在管腔和三阴性乳腺癌中抵抗抗性的新的治疗策略的观点。例如,通过将卡培他滨和他莫昔芬与药物(例如脱甲基酶抑制剂)的治疗相结合来防止在抗性细胞中观察到诸如h3k27me3等抑制性染色质标记的丢失可以是考虑使抗性最小化的策略。此外,在抗性样细胞中耗竭了转录抑制标记h3k27me3的基因座,包括已知可促进对化学疗法或靶向治疗的抗性的基因,突出了发现用于患者分层的新药物靶标和生物标记物的潜力。有趣的是,这两个模型在hgf信号通路中均具有h3k27me3-去甲基化和igfbp家族基因的转录激活,这在乳腺癌和抗药性中起关键作用。
在该实例中,对他莫昔芬的获得性抗性、scrna-seq和scchip-seq的模型独立地揭示了敏感肿瘤中具有抗性细胞共有的非遗传特征的一部分细胞的存在。两种方法没有揭示相同基因子集,但揭示了从管腔细胞向基底细胞身份的转变的共同基因特征。这样的差异可突出当前单细胞rna-seq方法的不良灵敏度,或者揭示了一部分基因准备转录但未被转录。抑制性染色质标记(例如h3k27me3)的丢失可将染色质更改为允许状态,并且可对应于转录变化之前的引发事件。
- 还没有人留言评论。精彩留言会获得点赞!