复合有机化合物的卤化
相关申请的交叉引用
本申请根据35u.s.c.§119(e)要求于2018年7月18日提交的美国临时申请第62/700,152号的权益,所述申请的公开内容通过引用以其整体并入本文。
政府支持声明
本发明是在美国国立卫生研究院(nationalinstitutesofhealth)授予的ca047135、ca070375和r01086374以及美国国家科学基金会(nationalsciencefoundation)授予的che-1205646的政府支持下完成的。政府拥有本发明的某些权利。
通过引用并入电子提交的材料
作为本公开的一部分的序列表与说明书作为文本文件同时提交。含有序列表的文本文件的名称为“53295a_seqlisting.txt”,所述文件于2019年7月18日创建,并且大小为84,638字节。序列表的主题通过引用以其整体并入本文。
本公开总体上涉及复合化学领域,并且更具体地涉及化合物卤化的生物催化。
背景技术:
卤化天然产物的流行已经促进了在了解参与次级代谢的各种类型的卤化酶方面的重大进步。到目前为止,大多数表征的卤化酶可以分为三类:卤过氧化物酶(含血红素且含钒)、非血红素fe(ii)α-酮戊二酸依赖性酶和黄素依赖性酶。卤过氧化物酶通常是非选择性的并通过利用自由扩散的次卤酸的机制执行卤化。相比之下,fe(ii)α-酮戊二酸依赖性卤化酶通过自由基机制,通常为卤化脂肪族未活化的碳1进行。黄素依赖性卤化酶(fdh)也通过次卤酸中间体进行,其中反应性试剂被表现为控制芳香族底物2,3上的卤化的区域选择性的赖氨酸残基捕获。fdh衍生的次卤酸是通过黄素c4a-过氧化物加合物与结合的氯离子之间的反应生成的。fdh被认为是通过亲电芳香族取代(eas)进行的,其中催化赖氨酸残基提供氯胺卤化剂,并且催化谷氨酸通过使在催化2期间产生的带正电的中间体去质子化来促进反应。
大部分先前表征的fdh是细菌来源的,其中从真核生物中报告的相对较少4-12,并且仍然以生物化学表征的更少4-6,10-12。已经发现细菌fdh在天然产物生物合成中催化游离的底物2,13-15和结合载体蛋白的底物16,17(包含前体氨基酸)两者上的反应。良好表征的真核生物fdhrdc24和chla6催化卤化代谢物的生物合成中的后期c-h官能化反应。然而,尚未报告这两种酶的结构数据,并且尚不清楚所述两种酶如何控制在大的、结构复合的底物上的位点选择性卤化。
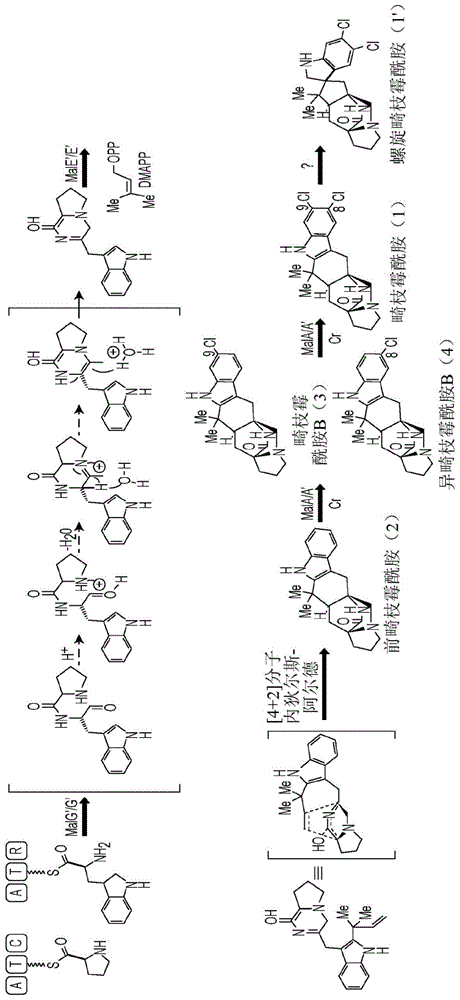
畸枝霉酰胺(malbrancheamide)(化合物1)是由陆地真菌金色畸枝霉(malbrancheaaurantiaca)rrc181318和海洋海绵衍生真菌禾本畸枝霉(malbrancheagraminicola)086937a19产生的复合的卤化吲哚生物碱。畸枝霉酰胺的发现是通过搜索钙调蛋白拮抗剂实现的,并且若干研究已经表征所述畸枝霉酰胺的显著的血管舒张作用20,21。从禾本畸枝霉19中分离出了近亲结构的螺旋畸枝霉酰胺(spiromalbramide)以及畸枝霉酰胺。这两个菌株是高度相关的,总体上具有99%的序列同一性,并且提出所述两个菌株的畸枝霉酰胺的生物合成途径是相同的(方案1)。畸枝霉酰胺(化合物1)属于通过以下形成的异戊烯基化吲哚生物碱的家族:非核糖体肽合成酶(nrps)的肽偶联、通过异戊烯基转移酶添加异戊二烯单元以及提出的[4+2]狄尔斯-阿尔德(diels-alder)环加成以形成前畸枝霉酰胺(化合物2)22-26的表征性[2.2.2]二氮杂辛烷环(方案1)。然后提出通过迭代机制对前畸枝霉酰胺进行二氯化,但是这种卤化是由一种还是两种卤化酶25执行尚未确定。吲哚环的氯化使此分子区别于其类别的剩余部分并且对所述分子的生物活性21具有显著贡献。
在阐明畸枝霉酰胺的生物合成途径的早期努力中,在金色畸枝霉中执行前体掺入研究。由此得出结论,前畸枝霉酰胺(premalbrancheamide)(化合物2)确实掺入一氯化畸枝霉酰胺b(化合物3)中,并且两种化合物均为金色畸枝霉的天然代谢物25。先前已经提出在c8官能化以产生畸枝霉酰胺(化合物1)25之前,对c9位置进行位点选择性氯化。然而,使c8(异畸枝霉酰胺(isomalbrancheamide)b(化合物4))和c9(畸枝霉酰胺b(化合物3))单卤化代谢物与金色畸枝霉20和禾本畸枝霉19分离与提出的c9选择性假设相冲突。
通过合成方法选择性地卤化高度复合分子中的c-h键的能力由于化学上等效的c-h键的丰度以及无法克服固有的空间或电子反应性偏向31,32而提出了巨大的挑战。通过定制酶来进行后期官能化的大量生物活性天然产物为利用卤化酶的能力执行困难的化学转化提供了独特的机会。
鉴于上述观察,本领域仍然需要修饰复合化合物的催化剂,例如如吲哚生物碱等复合化合物的受控卤化。
技术实现要素:
本公开提供了作为真菌mala或mala'卤化酶的氨基酸变体的具体可用于使如吲哚生物碱等复合化合物卤化的黄素依赖性卤化酶(fdh)。这些酶可用于在真菌畸枝霉酰胺途径中产生多种卤化化合物,所述化合物在哺乳动物中具有生理作用,如抑制钙调蛋白,并由此调节一系列多样的生理途径中涉及的钙离子信号传导途径,并且因此调节各种疾病或病症。包括施用有效量的本文所公开的化合物的示例性方法包含抑制哺乳动物中的平滑肌收缩。
在一方面,本公开提供了一种黄素依赖性卤化酶(fdh)变体,其与seqidno:2或seqidno:4中所示的氨基酸序列相比包括一个或两个氨基酸取代,其中所述fdh变体能够催化复合有机化合物的卤化。在一些实施例中,所述复合有机化合物是芳香族杂环有机化合物。在一些实施例中,所述芳香族杂环有机化合物包括双环[2.2.2]二氮杂辛烷环。在一些实施例中,所述芳香族杂环有机化合物包括吲哚。在一些实施例中,所述化合物是吲哚生物碱。在一些实施例中,所述吲哚生物碱是异戊烯基化吲哚生物碱。在一些实施例中,所述fdh变体源自真菌fdh。在一些实施例中,所述fdh变体源自细菌fdh。在一些实施例中,所述fdh变体不源自真菌fdh或细菌fdh。
在一些实施例中,所述fdh变体是mala卤化酶变体。在一些实施例中,所述fdh变体包括seqidno:2中所示的序列的氨基酸取代变体,其中取代为s129z、h253z、s129z/h253z、d109z、f489z、s409z、w265z、w263z、s82z、s129z/g131z、g131z、s129z/i493z、i493z、s129z/p85z或p85z,其中z是在指示位置处不存在氨基酸或存在除了野生型氨基酸之外的任何常规氨基酸。在本公开的上下文中,处于氨基酸水平的取代突变或变异被明确定义为包含任何天然存在的常规氨基酸对不相同氨基酸的取代,或无氨基酸对给定氨基酸的取代,即,单个氨基酸缺失。在一些实施例中,所述fdh变体包括seqidno:2中所示的序列的氨基酸取代变体,其中取代为s129a、h253f、s129a/h253f、h253a、d109a、f489h、s409a、w265a、w263a、s82a、s129a/g131s、g131s、s129a/i493s、i493s、s129a/p85s或p85s。在一些实施例中,所述mala卤化酶包括seqidno:6、8、12或16中所示的序列。在一些实施例中,所述fdh变体是mala'卤化酶变体。在一些实施例中,所述fdh变体包括seqidno:4中所示的序列的氨基酸取代变体,其中取代是e494z或h253z,其中z是在指示位置处不存在氨基酸或存在除了野生型氨基酸之外的任何常规氨基酸。在一些实施例中,所述fdh变体包括seqidno:4中所示的序列的氨基酸取代变体,其中取代为e494d、h253f、s129a、s129a/i493s、i493s、s129a/p85s、p85s、s129a/g131s或g131s。在一些实施例中,所述mala'卤化酶包括seqidno:10、14或18中所示的序列。
本公开的另一方面涉及一种多核苷酸,其对本文所公开的fdh变体进行编码。在一些实施例中,经过编码的fdh变体是mala卤化酶变体或mala'卤化酶变体。在一些实施例中,所述经过编码的fdh变体包括seqidno:6、8、10、12、14、16或18中所示的序列。
本公开的又另一方面涉及一种载体(vector),其包括本文所公开的多核苷酸。本公开的相关方面是一种宿主细胞,其包括本文所公开的多核苷酸或本文所公开的载体。
本公开的另一方面是一种畸枝霉酰胺d化合物,其包括式i:
本公开的相关方面是一种异畸枝霉酰胺d化合物,其包括式ii:
本公开的相关方面是一种畸枝霉酰胺e化合物,其包括式iii:
本公开的仍另一方面是一种使复合有机化合物卤化的方法,所述方法包括在适于对复合有机化合物进行酶催化卤化的条件下使所述复合有机化合物与本文所公开的fdh卤化酶变体以及卤素接触。在一些实施例中,所述复合有机化合物是芳香族杂环有机化合物。在一些实施例中,所述芳香族杂环有机化合物包括双环[2.2.2]二氮杂辛烷环。在一些实施例中,所述芳香族杂环有机化合物包括吲哚。在一些实施例中,所述化合物是吲哚生物碱。在一些实施例中,所述吲哚生物碱是异戊烯基化吲哚生物碱。在一些实施例中,所述异戊烯基化吲哚生物碱源自畸枝霉属。在一些实施例中,所述畸枝霉属是金色畸枝霉或禾本畸枝霉。在所述方法的一些实施例中,所述异戊烯基化吲哚生物碱是前畸枝霉酰胺、畸枝霉酰胺b、异畸枝霉酰胺b、畸枝霉酰胺c或异畸枝霉酰胺c。在一些实施例中,所述卤化步骤是氯化步骤。在一些实施例中,所述卤化步骤是溴化步骤。
本公开的另一方面涉及一种在体外调节哺乳动物的细胞或细胞中的ca2+信号传导途径的方法,所述方法包括在体外向所述哺乳动物、所述细胞或在体外向一种或多种分离酶施用有效量的卤化复合有机化合物。一些实施例实施一种调节哺乳动物的细胞中的ca2+信号传导途径的方法,所述方法包括向所述哺乳动物施用有效量的卤化复合有机化合物。在一些实施例中,所述ca2+信号传导途径是ca2+钙调蛋白依赖性途径。在一些实施例中,调节所述ca2+信号传导途径抑制平滑肌收缩。在一些实施例中,所述卤化复合有机化合物是畸枝霉酰胺、畸枝霉酰胺b、异畸枝霉酰胺b、畸枝霉酰胺c、异畸枝霉酰胺c、畸枝霉酰胺d、异畸枝霉酰胺d或畸枝霉酰胺e。
一些实施例实施一种在体外调节细胞(如哺乳动物细胞)中或在体外调节一种或多种分离酶中的ca2+信号传导途径的方法,所述方法包括在体外向所述细胞或在体外向所述一种或多种分离酶施用有效量的卤化复合有机化合物。一些实施例提供了一种在体外调节细胞中的ca2+信号传导途径的方法,所述方法包括在体外向所述细胞施用有效量的卤化复合有机化合物。在一些实施例中,所述ca2+信号传导途径是ca2+钙调蛋白依赖性途径。在一些实施例中,在体外调节的所述ca2+信号传导途径参与体内平滑肌收缩。在一些实施例中,所述卤化复合有机化合物是畸枝霉酰胺、畸枝霉酰胺b、异畸枝霉酰胺b、畸枝霉酰胺c、异畸枝霉酰胺c、畸枝霉酰胺d、异畸枝霉酰胺d或畸枝霉酰胺e。一些实施例实施一种调节ca2+信号传导途径的方法,所述方法包括向一种或多种分离酶施用有效量的卤化复合有机化合物。在一些实施例中,所述ca2+信号传导途径是ca2+钙调蛋白依赖性途径。在一些实施例中,在体外调节的所述ca2+信号传导途径参与体内平滑肌收缩。在一些实施例中,所述卤化复合有机化合物是畸枝霉酰胺、畸枝霉酰胺b、异畸枝霉酰胺b、畸枝霉酰胺c、异畸枝霉酰胺c、畸枝霉酰胺d、异畸枝霉酰胺d或畸枝霉酰胺e。
通过参考以下详细描述(包含附图和实例),将更好地理解本公开的其它特征和优点。
附图说明
本专利或申请文件含有至少一幅彩色附图。在请求并支付必要的费用后,将由美国专利商标局(unitedstatespatentandtrademarkoffice)提供带有一幅或多幅彩色附图的本专利或专利申请出版物的副本。
图1.方案1.金色畸枝霉和禾本畸枝霉中的畸枝霉酰胺生物合成途径,其中螺旋畸枝霉酰胺仅在禾本畸枝霉中产生。
图2.金色畸枝霉和禾本畸枝霉中的畸枝霉酰胺生物合成基因簇。
图3.畸枝霉酰胺和相关代谢物。金色畸枝霉分离物包含化合物1、2、3和4,而禾本畸枝霉还产生化合物5和6。化合物7和8先前未从任一种生物体中描述过。有关化合物身份,参见图1。
图4.mala体外反应的hplc痕量(240nm),(a)与来自真菌提取物(i)的标准品相比的化合物2的氯化(见图1)(ii)、(b)与无酶对照物(nec)(i)相比的化合物2的溴化(ii)以及(c)与nec(i)相比以及化合物3的溴化(iv)与nec(ii)相比的化合物4的溴化(iii)。有关化合物身份,参见图1。
图5.吲哚区的ghmbcad和gcosynmr关联,用于测定新的畸枝霉酰胺类似物的卤化的位点。
图6.复合物的mala'活性位点与底物2(黄色)、3(粉色)和4(青色)的重叠。有关底物或化合物2、3和4的身份,参见图1。
图7.mala'的活性位点揭示底物与fad结合袋之间的明显分离。
图8.在与化合物2反应以产生化合物4(绿色)、3(蓝色)和1(红色)时,突变体对野生型mala的转化率百分比。malak108a和e494a/q非活性,并且malas82a不溶于水且未进行功能分析。有关化合物身份,参见图1。
图9.mala'phe489(橙色)与底物(a)前畸枝霉酰胺(化合物2)、(b)畸枝霉酰胺b(化合物3)和(c)异畸枝霉酰胺b(化合物4)之间的相互作用。有关化合物身份,参见图1。
图10.与化合物2共结晶的野生型mala'(黄色)和mala'e494d(青色)的比较。有关化合物身份,参见图1。
图11.与化合物2共结晶的野生型mala'(黄色)和mala'h253f(青色)的比较。有关化合物身份,参见图1。
图12.a)取自对mala'和底物2结合复合物(包含lys108处的氯胺加合物)进行的500纳秒md模拟的代表性快照(40纳秒处)。b)针对以下三种模型计算的dft反应途径:1a-吲哚和甲基氯胺;分别在c8或c9位置处配位的1b-吲哚、甲基氯胺和水分子;与h-c8配位的1c-吲哚、甲基氯胺和meoh。c)dft优化了一些代表性结构的几何形状。吉布斯能以kcal/mol的形式给出,并且距离以
图13.方案2.提出的用于mala催化的机制。
图14.表征和未表征(用生产生物体标记)的fdh的系统发育树。基于序列比对,与蓝色区中的mala簇具有高氨基酸同一性(大于50%)的推定真菌fdh预期含有zn2+结合基序。绿色区含有先前表征的细菌fdh(真核chla除外),并且黄色区表示来自真菌的推定fdh的新亚类。使用lasergenemegalignpro(dnastar)40制备的系统发育树。
图15.mala变体表现出对天然底物前畸枝霉酰胺的一系列位点选择性。可以移动底物位置以实现位点选择性。
图16.与黑色突变体(a)mala'(malal276p/r428p)、(b)malaw263a、(c)malaw265a、(d)malas409a、(e)malah253a、(f)malaf489h相比的野生型mala(粉色)反应。所述反应由mala或mala变体结合非特异性黄素还原酶、过量的黄素辅因子、作为氯离子源的nacl和nadh辅因子构成。通过hplc监测240nm处的吸光度来执行分析。
图17.与(b)malad109a和(c)malas129a相比的(a)野生型mala反应。所述反应由mala或mala变体结合非特异性黄素还原酶、过量的黄素辅因子、作为氯离子源的nacl和nadh辅因子构成。通过hplc监测240nm处的吸光度来执行分析。
图18.与(b)malas129a、(c)malah253f和(d)malas129a/h253f相比的(a)野生型mala反应。所述反应由mala或mala变体结合非特异性黄素还原酶、过量的黄素辅因子、作为氯离子源的nacl和nadh辅因子构成。通过hplc监测240nm处的吸光度来执行分析。
图19.化合物5和6的氯化以产生化合物8和7。顶部(a)标准化合物5、(b)标准化合物8、(c)对照化合物5、(d)化合物5的氯化反应。底部(a)标准化合物6、(b)标准化合物7、(c)对照化合物6、(d)化合物6的氯化反应、(e)化合物2的氯化反应以产生化合物1标准品。
图20.与野生型mala相比,化合物2与malah253a和malah253f的溴化。(a)标准化合物2、(b)标准化合物5、(c)标准化合物6、(d)与化合物2进行的wtmala溴化反应、(e)与化合物2进行的malah253a溴化反应以及(f)与化合物2进行的malah253a溴化反应。
图21.dft使用以下三种计算模型优化了c8和c9氯化的反应途径:a)吲哚环和甲基氯胺;b)分别更靠近c8-或c9-h的吲哚环、甲基氯胺和水分子;c)作为ser129模型的吲哚环、甲基氯胺和甲醇分子。在m06-2x/6-311+g(d,p)/cpcm(乙醚)//m06-2x/6-31g(d)/cpcm(乙醚)水平上计算相对吉布斯自由能(δg,以kcal/mol计)。键长以
具体实施方式
真菌属畸枝霉包含如金色畸枝霉属和禾本畸枝霉等含有用于产生如卤化吲哚生物碱等复合卤化化合物的生物合成途径的物种,所述吲哚生物碱具有有用的生理作用,如可用于调节哺乳动物细胞中的参与许多生理功能并且涉及多种疾病和病症的钙离子信号传导途径。本公开提供了如金色畸枝霉的mala卤化酶和禾本畸枝霉的mala'卤化酶等真菌卤化酶的变体,所述卤化酶催化可用于例如抑制平滑肌收缩的复合卤化化合物的合成。数据提供了对真菌卤化酶变体的全面表征并提供了用作底物或由卤化酶变体产生的化合物的结构和性质的广泛表征,这些化合物通常属于畸枝霉真菌属的畸枝霉酰胺合成途径。
本文所公开的实验被设计为鉴别和表征畸枝霉酰胺生物合成中涉及的通用卤化酶,并且证明其作为用于复合有机化合物的卤化的生物催化剂的潜力,所述复合有机化合物包含在畸枝霉酰胺途径中发现的各种化合物,如前畸枝霉酰胺(化合物2)和单卤化和双卤化畸枝霉酰胺途径化合物两者,包含畸枝霉酰胺b(化合物3)、异畸枝霉酰胺b(化合物4)、畸枝霉酰胺c、异畸枝霉酰胺c、畸枝霉酰胺d和异畸枝霉酰胺d(参见图1和3)。实验探讨了吲哚环体系上前畸枝霉酰胺在两个相邻位置处的迭代后期卤化的机制。使c8(异畸枝霉酰胺b(化合物4))和c9(畸枝霉酰胺b(化合物3))单卤化代谢物与金色畸枝霉20和禾本畸枝霉19分离与提出的c9选择性假设相冲突,从而为研究畸枝霉酰胺卤化过程提供了另外的动力。
对金色畸枝霉和禾本畸枝霉进行的基因组测序和生物信息学分析导致分别鉴别mala和mala'。这两种fdh同一性为99%,仅两个氨基酸不同,并且被提出作为每个生物体26的畸枝霉酰胺生物合成途径中的最后一步来催化双卤化(图2)。来自含有nrps的基因簇的黄素依赖性卤化酶对游离底物进行的后期卤化是不常见的。卤化通常作为细菌非核糖体肽生物合成1中的第一步发生在激活氨基酸之前。在先前表征的黄素依赖性卤化酶中,大多数作用于如单个氨基酸2,13-15,27或载体蛋白束缚的小分子16,17,28,29等亚基底物。就复合多环底物的后期活性而言,与mala最接近的比较是源自蓝藻细菌的welo5非血红素fe(ii)α-酮戊二酸依赖性卤化酶,所述卤化酶作用于费希尔吲哚(fischerindole)和卤化吲哚(hapalindole)生物碱30。除了底物范围分析之外,还已经探索了黄素依赖性卤化酶中的卤素选择性,并且发现大多数卤化酶催化氯化反应和溴化反应两者。在海洋真菌菌株禾本畸枝霉中使用高浓度溴盐的前体掺入研究中,前畸枝霉酰胺(化合物2)的溴化产生畸枝霉酰胺c(5)和异畸枝霉酰胺c(化合物6)19(图3)。
畸枝霉酰胺是从金色畸枝霉和禾本畸枝霉两者中分离得到的二氯化真菌吲哚生物碱,所述生物碱属于含有表征性双环[2.2.2]二氮杂辛烷核的天然产物的家族。在畸枝霉酰胺的吲哚环上引入氯原子使其与此家族的其它成员区分开并且对其生物学活性作出了重要贡献。已经表征了在两种不同的真菌菌株中的参与畸枝霉酰胺的后期卤化的两种黄素依赖性卤化酶。作为畸枝霉酰胺生物合成途径中的最终步骤,mala和mala'催化游离底物前畸枝霉酰胺的迭代二氯化和单溴化。通过mala介导的化学酶合成生成两种非天然的溴-氯-畸枝霉酰胺类似物。与三种底物复合的mala'的结构分析和计算研究揭示,酶代表一类新的锌结合黄素依赖性卤化酶并且为预期独特的反应机制提供了新见解。
本文所公开的实验结果提供了黄素依赖性卤化酶的独特亚类的实例,所述亚类执行独立于载体蛋白的复合底物的迭代后期卤化。mala编码在含有非核糖体肽合成酶(nrps)的基因簇中,但本文提供的证据证明,此蛋白催化游离底物上的有效的后期官能化。数据带来了对涉及ser129的用于在mala/mala'卤化中使带正电的韦兰德(wheland)中间体去质子化(如图13所示的方案2)的新机制的预期。glu494与吲哚氮之间的氢键预期会增加芳香族环的亲核性。这促进了亲电芳香族取代(eas)反应,从而产生带正电荷的中间体。然后,水分子或丝氨酸侧链可以使韦兰德中间体去质子化,从而造成吲哚环体系的重新芳构化(方案2,图13)。
如以下实例所揭示的,活性位点中的两个关键残基对底物结合即glu494和phe489具有显著贡献。glu494与吲哚氮和phe489的氢键促进了与吲哚的芳香族环的有利疏水相互作用。相对于野生型mala,malaf489h的活性显著降低,尤其是对于第二次氯化反应而言。预期活性位点背面的苯丙氨酸残基有助于底物结合并保持与一氯化产物的相互作用,以促进第二次卤化反应。
米-曼氏模型动力学(michaelis-mentenmodelkinetics)在化合物2的c9位点和c8位点两者处表现出相等的一氯化速率,并且表现出化合物3和4的相等的氯化速率,从而得出mala对于吲哚环的两个位点的选择性相等的结论。有趣的是,第二次氯化氯化的催化效率(kcat/km)值是第一次氯化观察到的催化效率值的两倍,因此,预期初始氯化会为第二次卤化提供底物。这种作用可以与结构数据相关,其中吲哚底物上的氯原子与phe489有利地相互作用,从而可能促进更好的结合模式。另外,二氯化畸枝霉酰胺(化合物1)未结合在晶体中,这表明二氯化的解离速率比易于结合在mala'晶体中的一氯化产物中的任一种产物的解离速率都快。
为了阐明在mala中的选择性的机制,使用催化赖氨酸附近的组氨酸残基来探测活性位点区。malah253a表现出对化合物2的c9位置处的氯化的选择性,而malah253f对化合物2的c8位置具有选择性。md模拟和dft计算证明了活性位点中的极性氨基酸侧链(ser129)或水分子与吲哚的不同c-h位置之间的关键相互作用如何控制对氯化反应的选择性。这可以通过在eas期间增强碳原子的亲核性来实现,但也可以通过预先组织碱基以执行最终的去质子化步骤来实现。his253处的丙氨酸取代阻止了ser129与h-c8的相互作用,从而产生有利于c9处的氯化的整个非极性活性位点环境。另一方面,可以通过与h-c8的更有效的ser129相互作用来解释在malah253f中观察到的c8选择性。当在溴化反应的位点选择性方面研究这些突变体时,观察到野生型产物概况。与hocl相比,hobr是较温和的卤化试剂,因此有利于化合物2固有的反应性更高的c9位点。这些发现证明,甚至是对活性位点区进行很小的调节也可能导致卤化反应的位点选择性的重大变化。
将mala指定为一类新的黄素依赖性卤化酶不仅通过其反应性,而且还通过其独特的结构基序得到例证:zn2+结合c末端和扩展的活性位点能够容纳复合底物。此zn2+结合基序的发现提供了用于挖掘mala同系物的序列数据以寻求用于后期卤化的生物催化剂的指纹(图13)。对mala/mala'的研究已经为复合底物的迭代后期卤化的生物催化机制提供了新见解。
以下实例通过图示方式呈现并且不旨在限制本文公开的主题的范围。
实例
实例1
材料和方法
禾本畸枝霉基因组dna提取和测序
将丝状真菌菌株禾本畸枝霉在静态100ml马铃薯葡萄糖肉汤(pbd)培养基上在26℃下培养10天。基因组dna(gdna)提取方案和测序方案与li等人1中使用的方案相同,并且使用索雷克萨公司(solexa)测序以测定基因组序列。
金色畸枝霉cdna制备
在以160rpm振荡的pdb中在28℃下培养金色畸枝霉持续15天。英杰公司(invitrogen)purelinkrna迷你试剂盒与来自相关rneasy迷你手册(2010)中的植物和真菌组织处理方案一起使用,以在用dnase处理前分离rna。将英杰公司superscript第一链合成与protoscriptm-mulv第一链cdna合成试剂盒和方案一起使用以生成cdna。使用以下引物和以下pcr循环,通过pcr从cdna模板扩增mala编码区:(1)94℃持续2分钟,(2)98℃持续10秒,(3)66.3℃持续30秒,(4)68℃持续2分钟,步骤2-4重复40次。
引物
5'-gagagctagcatggcgccgacaccaaagtatacgt-3'(seqidno:19)
5'-cattaagcttctatgcagctggcctggtaggggtt-3'(seqidno:20)
mala-pmcsg7和mala'-pmcsg7的克隆
将mala-pcr产物通过连接非依赖性克隆(lic)2插入pmcsg7载体中。用mala-pmcsg7转化大肠杆菌xl1蓝细胞以进行筛选和质粒保持。mala'-pmcsg7是通过如下所述的定点诱变制备的。已经对hpac黄素还原酶(phac质粒)3进行了描述。
金色畸枝霉生长和畸枝霉酰胺的提取
分离和纯化程序改编自martínezluis等人4。将75ml马铃薯葡萄糖肉汤的单独烧瓶与金色畸枝霉的100μl孢子储备液接种并生长三周,或直到产生白色真菌垫。在明显的橙色孢子形成之前,将培养物粉碎并且用二氯甲烷提取。首先用1mhcl对粗提取物进行酸碱纯化,然后用2m氢氧化铵中和到ph9,并且用二氯甲烷进行反提取。然后在飞诺美(phenomenex)lux5μm纤维素-3250×10mm柱上通过手性hplc纯化提取物。使用以下hplc时间程序对畸枝霉酰胺化合物进行分离和纯化:50%乙腈持续18分钟,在2分钟内梯度到55%乙腈、55%乙腈持续2分钟,在2分钟内梯度到40%乙腈、40%乙腈持续5分钟,流速为4毫升/分钟。流动相由水和乙腈组成。从金色畸枝霉的1.5l生长中获得以下天然存在的畸枝霉酰胺的产量:1.6mg/l前畸枝霉酰胺(化合物2)(1h-nmr,400mhz,cd3od,
mala表达和纯化
mala、mala'和mala/a'突变体的表达。用mala-pmcsg7和来自塔卡拉公司(takara)的pgro7分子伴侣质粒组(groel/groes)转化大肠杆菌菌株bl21(de3)。将氨苄青霉素(0.1mg/ml)、氯霉素(35μg/ml)和l-阿拉伯糖(0.5mg/ml)加入到1l极品肉汤(terrificbroth,tb)培养基中,然后用转化的大肠杆菌细胞接种。使1l培养物在37℃下生长,直到od600达到0.8-1.0,在20℃下冷却一小时,用0.1mm异丙基β-d-1-硫代半乳糖吡喃糖苷(iptg)诱导并且20℃下表达18小时。
mala'对于硒代甲硫基mala'的表达。向450ml硒代蛋氨酸培养基(athenaes)补充25mltb培养基和150μg/ml硒代-dl-蛋氨酸。将氨苄青霉素(0.1mg/ml)、氯霉素(35μg/ml)和l-阿拉伯糖(0.5mg/ml)加入到培养基中,然后用转化的大肠杆菌细胞接种。使细胞培养物在37℃下生长,直到od600达到0.6,在20℃下冷却一小时,用0.1mmiptg诱导并在20℃下表达18小时。
用于氯化测定和大规模反应的蛋白质纯化。将来自500ml培养物的细胞团粒重悬于30ml裂解缓冲液nacl(10%(v/v)甘油、500mmnacl、ph为7的20mm咪唑、ph为7的20mmhepes)中。向细胞悬浮液补充50μm黄素腺嘌呤二核苷酸(fad),并且用5mg溶菌酶、2mgdnase和3mmmgso4使细胞裂解。细胞裂解通过超声处理完成,并且细胞废物在标准细胞碎片造粒条件下通过离心清除(例如,39,200rcf下持续25分钟)。过滤上清液并且通过金属亲和色谱法在5mlhis-trap柱(通用电气医疗公司(gehealthcare))上纯化mala,其中洗脱缓冲液nacl(10%甘油、500mmnacl、ph为7的30-560mm咪唑、ph为7的20mmhepes)的体积梯度为10柱。蛋白质在冰上用2mmatp和50μmfad温育并且在superdexs20016/60hiload柱上使用存储缓冲液nacl(10%甘油、300mmnacl、ph为7的20mmhepes)通过大小排阻色谱法进一步进行纯化以去除伴侣蛋白。每1l的细胞培养物获得20mg纯化的mala。
用于溴化测定和大规模反应的蛋白质纯化。将来自500ml表达培养物的细胞团粒重悬于30ml裂解缓冲液nabr(50mmnah2po4、ph为7的10mm咪唑、300mmnabr、10%甘油)中并补充有50μmfad。细胞裂解是通过加入5mg溶菌酶、2mgdnase和3mmmgso4以及超声处理完成的。在标准细胞碎片造粒条件下通过离心清除细胞废物(例如,39,200rcf下持续25分钟),并且通过与10mlni-nta超流树脂(凯杰公司(qiagen))分批结合以使蛋白质纯化。用洗涤缓冲液nabr(50mmnah2po4、ph为7的20mm咪唑、300mmnabr、10%甘油)洗涤树脂结合的蛋白质,并且用洗脱缓冲液nabr(50mmnah2po4、ph为7的250mm咪唑、100mmnabr、10%甘油,0.2mmtcep)使蛋白质洗脱。将溴化物结合的mala交换到pd-10柱(通用电气医疗公司)上的存储缓冲液nabr(50mmnah2po4、1mmedta、0.2mmdtt,10%甘油,ph7.3)中。
用于晶体学的蛋白质纯化。纯化的初始步骤与用于氯化测定的mala纯化的步骤相同。在补充有50μmfad和2mmdtt的储存缓冲液nacl的过夜透析中用tev蛋白酶(1mg蛋白酶/50mgmala)切割his标签。通过金属亲和色谱法使无标签mala与tev蛋白酶和任何剩余的his6-mala分离,并且用储存缓冲液nacl通过大小排阻色谱法使mala纯化。每1l的细胞培养物获得10mg纯化的mala。
mala生化活性测定
在具有以下组分的100μl体积中执行生化活性测定:18μmmala、54μmhpac黄素还原酶、3100μmfad、50mmnacl、250μm底物、5mmnadh,并且用反应缓冲液(与储存缓冲液nabr相同)调到总体积。氯化反应进行20分钟,并且溴化反应过夜进行。用乙酸乙酯(200μl,一式三份)提取反应物并且在氮气下使反应物干燥。经干燥提取物重悬于lc/ms级甲醇中以达到约10μm的浓度,以进行lc/ms分析。在安捷伦(agilent)四极杆飞行时间光谱仪(q-tof6500系列)上使用电喷雾电离执行高分辨率质谱分析。使用乙腈和水通过以下hplc方法监测生化活性:65%乙腈持续5分钟,在10分钟内梯度到95%乙腈、95%乙腈持续5分钟,在2分钟内梯度到65%乙腈、65%乙腈持续11分钟,以0.3毫升/分钟的流速重新平衡,在飞诺美lux纤维素-3、纤维素三(4-甲基苯甲酸酯)250×4.6mm柱上在240nm处监测。
mala'的共晶体结构
结晶条件。来自金色畸枝霉的mala难以结晶,而来自禾本畸枝霉的mala'(通过mala:l276p/r428p的定点诱变产生)被证明是最适合结晶。将纯化的mala'在具有200mmnacl或300mmnacl的ph为7的20mmhepes缓冲液中透析过夜,以去除甘油,并且然后补充有等摩尔量的fad。将mala'与等摩尔浓度的异畸枝霉酰胺b(化合物4)进行预温育得到具有结合在晶格接触而非活性位点中的化合物4的晶体。对于具有化合物2、3和4的活性位点复合物,用四倍摩尔过量的底物预温育mala'。通过蒸汽扩散法从用化合物2、3或4预温育的5mg/mlmala'与含有2m(nh4)2so4、0.2mli2so4、5mmcdcl2和ph为5.5的0.1mbis-tris的孔溶液的1:2混合物中生长晶体。晶体在增添10%甘油的孔溶液中冷冻保护,并且在液氮中快速冷却。
数据收集。在阿贡国家实验室(argonnenationallaboratory)的先进光子源(aps)处的gm/ca光束线23id-b处收集数据。对于semet-mala'晶体,使用30°楔形件以反向光束几何形状收集180°衍射数据。所有数据均使用xds5处理。使用phenix套件中的autosol通过单波长异常衍射(sad)对semetmala'卤化酶结构进行解析,以定位se位点,确定初始相并且执行密度修饰(品质因数=0.236)6。phenix套件中的autobuild用于构建82%完整的启动模型。semetmala模型用作分子替代中的模板,以使用phenix套件中的phaser对天然mala结构进行解析。用七个平移/旋转(libation)/螺旋组7使用coot和phenixrefine进行模型构建和优化进程,以完成模型。
定点诱变
mala'(malal276p/r428p)。从r428p开始,依次执行定点诱变(sdm)以制备l276p/r428p双重取代。反应包含100ngmala-pmcsg7模板、每个引物(正向l276p和反向l276p)100ng、5μl10×pfu缓冲液、0.5μldntp(每个250μm)和1μl来自安捷伦的pfuturbo(总计50μl)。pcr循环为(1)95℃持续30秒,(2)95℃持续30秒,(3)55℃持续1分钟,(4)68℃持续8分钟;步骤2-4重复16个循环。含有0.5μl2u/μldpni和25μlpcr反应溶液的dpni消化在37℃下持续2小时,并且在通过碱性裂解(来自英杰公司的purelink快速质粒微量制备试剂盒)和测序进行的质粒分离之前执行,以验证突变体的存在。使用具有100ngmalar428p模板的单个引物sdm、0.2μm引物、250μmdntp、5μl10×pfu缓冲液、1μlpfu融合聚合酶(总体积为50μl)来制备l276p取代。pcr时间程序如下:(1)95℃持续3分钟,(2)95℃持续35秒,(3)52℃持续50秒,(4)65℃持续13分钟,(5)65℃持续15分钟;步骤2-4重复30个循环。以与上述相同的方式执行dpni消化和序列分析。
引物
r428pforward5'-gcacagctttcgcacccaattgtggagattggg-3'(seqidno:21)
r428preverse5'-cccaatctccacaattgggtgcgaaagctgtgc-3'(seqidno:22)
l276p5'-cgtctaccctcttgggaagggagccccatagcgaacttgatggatatgg-3'(seqidno:23)
malak108a。使用quikchange闪电定点诱变试剂盒和方案制备malak108a突变体。所使用的pcr时间程序为(1)95℃持续2分钟,(2)95℃持续20秒,(3)55℃持续30秒,(4)65℃持续6分钟,(5)65℃持续5分钟;步骤2-4重复30个循环。qcldpni消化和转化方案与xl10-金超感受态细胞一起使用。
引物
5'-gtaaaagcacagcccatccgcgagtccgaatagtcgaagg-3'(seqidno:24)
所有其它mala/mala'突变体。使用具有100ngmala或mala'模板的单个引物sdm、0.2μm引物、250μmdntp、5μl10×pfu缓冲液、1μlpfu融合聚合酶(总体积为50μl)来制备突变体。pcr时间程序如下:(1)95℃持续3分钟,(2)95℃持续35秒,(3)x℃(参见下文)持续50秒,(4)65℃持续13分钟,(5)65℃持续15分钟;步骤2-4重复30个循环。以与上述相同的方式执行dpni消化和序列分析。
引物
mala大规模反应和产物分离
氯化反应条件和提取。用90μmmala、54μmhpac黄素还原酶、250μm化合物2、100μmfad、50mmnacl、5mmnadh在1ml等分试样中运行反应,并且用反应缓冲液(与储存缓冲液nabr相同)调到总体积。20分钟后用2ml乙酸乙酯一式三份提取反应物,在氮气下干燥,并且重悬于甲醇中以进行hplc纯化。在5.1mg的反应中,分离出1.7mg畸枝霉酰胺b(化合物3)、1.3mg异畸枝霉酰胺b(化合物4)和1.2mg畸枝霉酰胺(化合物1)。
溴化反应条件和提取。用40μmmala、54μmhpac黄素还原酶、250μm化合物2、100μmfad、50mmnabr、5mmnadh在1ml等分试样中运行反应,并且用反应缓冲液(与储存缓冲液nabr相同)调到总体积。12小时后用2ml乙酸乙酯一式三份提取反应物,在氮气下干燥,并且重悬于甲醇中以进行hplc纯化。在用底物(化合物)2进行的3.7mg反应中,分离出0.9mg畸枝霉酰胺c(化合物5)和0.7mg异畸枝霉酰胺c(化合物6)。在用化合物3进行的2mg反应中,分离出480μg化合物7和300μg化合物1。在用化合物4进行的2.3mg反应中,分离出1.8mg化合物8。
hplc纯化。采用与用于使真菌提取物纯化的方法相同的手性hplc方法使畸枝霉酰胺b产物(化合物3)、异畸枝霉酰胺b产物(化合物4)和畸枝霉酰胺(化合物1)产物纯化。用以下hplc时间程序使用手性hplc与先前提及的半制备型纤维素柱分离出畸枝霉酰胺c产物(化合物5)、异畸枝霉酰胺c产物(化合物6)、畸枝霉酰胺d产物(化合物7)和异畸枝霉酰胺d(化合物8)产物:70%乙腈持续14分钟,在2分钟内以4毫升/分钟的流速梯度到60%乙腈。
米-曼氏模型动力学
底物畸枝霉酰胺b(化合物3)和异畸枝霉酰胺b(化合物4)产生畸枝霉酰胺(化合物1)。用以下组分在250μl的总体积中建立反应:1.1μmmala、44μmhpac黄素还原酶、100μmfad、50mmnacl、3.6mmnadh以及浓度在1μm到60μm范围的各种底物。通过在每个时间点(2、5、10、15分钟)去除50μl来用甲醇使反应淬灭。用以下lc时间程序在schimadzuhplc上分析反应:40%乙腈持续1分钟,在6分钟内从40%乙腈梯度到85%乙腈,85%乙腈持续1分钟,在1分钟内梯度到40%乙腈,重新平衡到40%乙腈持续3分钟。在240nm处测量吸光度,并且流动相由水和乙腈组成。采用飞诺美-lux纤维素-3、纤维素三(4-甲基苯甲酸酯)250×4.6mm柱进行分离。使用graphpadprism(6.01版)软件绘制初始速度相对于底物浓度并测定动力学常数kcat和km。
底物前畸枝霉酰胺(化合物2)产生异畸枝霉酰胺b(化合物4)和畸枝霉酰胺b(化合物3)。用以下组分在250μl的总体积中建立反应:1.8μmmala、44μmhpac、100μmfad、50mmnacl、3.6mmnadh以及浓度在5μm到80μm范围的各种底物。通过在每个时间点(2、5、7、10分钟)去除50μl来用100μl甲醇使反应淬灭。用以下lc时间程序在schimadzuhplc上分析反应:34%乙腈持续1分钟,在11分钟内梯度到62%乙腈,62%乙腈持续30秒,在30秒内梯度到34%乙腈,重新平衡到34%持续3分钟。在240nm处测量吸光度,并且流动相由水和乙腈组成。采用飞诺美-lux纤维素-3、三(4-甲基苯甲酸酯)250×4.6mm柱进行分离。
计算方法
dft计算。使用高斯09(修订版d.01)8执行dft计算。在cpcm可极化导体模型(乙醚,ε=4)10,11和6-31g(d)基组内,使用m06-2x9对所有几何形状进行优化。使用相同的dft泛函和溶剂化模型以及6-311++g(d,p)基组计算单点能量。使用所得能量校正从m06-2x/6-31g(d)优化12获得的气相能量。计算了1atm和298.15k的焓和熵。通过振动频率分析,将所有固定点验证为最小或一阶鞍点。介电常数ε=4的使用已经被证明是用于解释酶活性位点13,14中的电子极化和小骨架波动并且估计酶活性位点中的介电常数的很好的通用模型。用cylview15说明计算结构。
分子动力学模拟。使用amber16包17的gpu代码(pmemd)16执行分子动力学(md)模拟。使用一般amber力场(gaff)18在前室(antechamber)模块中生成中间体cl-k和底物的参数,其中将部分电荷设置为适合resp模型19在hf/6-31g(d)水平上产生的静电势。根据merz–singh–kollman方案20,21使用高斯09包8计算电荷。使用跳跃模块将每种蛋白质浸入具有tip3p22水分子的
仅实例1的参考文献
(1)li,s.;anand,k.;tran,h.;yu,f.;finefield,j.m.;sunderhaus,j.d.;mcafoos,t.j.;tsukamoto,s.;williams,r.m.;sherman,d.h.《药物化学通讯(med.chem.commun.)》2012,3,987-996。
(2)martínez-luis,s.;rodríguez,r.;acevedo,l.;gonzález,m.c.;lira-rocha,a.;mata,r.《四面体(tetrahedron)》2006,62,1817-1822。
(3)chakraborty,s.;ortiz-maldonado,m.;entsch,b.;ballou,d.p.《生物化学(biochemistry)》2010,49(2),372-385。
(4)stols,l.;gu,m.;diekman,l.;raffen,r.;collart,f.r.;donnelly,m.i.《蛋白表达和纯化(proteinexpressionandpurification)》2002,25,8-15。
(5)kabsch,w.;《晶体学学报d:生物晶体学(acta.crystallogr.d.biol.crystallogr.)》2010,66,125-132。
(6)adams,p.d.;afonine,p.v.;bunkoczi,g.;chen,v.b.;davis,i.w.;echols,n.;headd,j.j.;hung,l.w.;kapral,g.j.;grosse-kunstleve,r.w.;mccoy,a.j.;moriarty,n.w.;oeffner,r.;read,r.j.;richardson,d.c.;richardson,j.s.;terwilliger,t.c.;zwart,p.h.《晶体学学报d:生物晶体学》2010,66,213-221。
(7)emsley,p.;cowtan,k.《晶体学学报d:生物晶体学》2004,60,2126-2132。
(8)高斯09,修订版d.01,沃灵福德(wallingford)ct2013。frisch,m.j.;trucks,g.w.;schlegel,h.b.;scuseria,g.e.;robb,m.a.;cheeseman,j.r.;scalmani,g.;barone,v.;mennucci,b.;petersson,g.a.;nakatsuji,h.;caricato,m.;li,x.;hratchian,h.p.;izmaylov,a.f.;bloino,j.;zheng,g.;sonnenberg,j.l.;hada,m.;ehara,m.;toyota,k.;fukuda,r.;hasegawa,j.;ishida,m.;nakajima,t.;honda,y.;kitao,o.;nakai,h.;vreven,t.;montgomery,j.a.,jr.;peralta,j.e.;ogliaro,f.;bearpark,m.;heyd,j.j.;brothers,e.;kudin,k.n.;staroverov,v.n.;kobayashi,r.;normand,j.;raghavachari,k.;rendell,a.;burant,j.c.;iyengar,s.s.;tomasi,j.;cossi,m.;rega,n.;millam,m.j.;klene,m.;knox,j.e.;cross,j.b.;bakken,v.;adamo,c.;jaramillo,j.;gomperts,r.;stratmann,r.e.;yazyev,o.;austin,a.j.;cammi,r.;pomelli,c.;ochterski,j.w.;martin,r.l.;morokuma,k.;zakrzewski,v.g.;voth,g.a.;salvador,p.;dannenberg,j.j.;dapprich,s.;daniels,a.d.;farkas,
(9)zhao,y.;truhlar,d.g.《理论化学述评(theor.chem.acc.)》2008,120,215。
(10)barone,v.;cossi,m.《物理化学期刊a(j.phys.chem.a.)》1998,102,1995。
(11)cossi,m.;rega,n.;scalmani,g.;barone,v.《计算化学杂志(j.comp.chem.)》2003,24,669。
(12)simon,l.;goodman,j.m.《有机与生物分子化学(org.biomol.chem.)》2011,9,689。
(13)schutz,c.n.;warshel,a.《蛋白质:结构、功能与生物信息学(proteins:struct.funct.bioinf.)》2001,44,400。
(14)li,l.;li,c.;zhang,z.;alexov,e.《化学理论与计算(j.chem.theorycomp.)》2013,9,2126。
(15)legault,c.加拿大魁北克舍布鲁克舍布鲁克大学(universitédesherbrooke,sherbrooke,quebec,canada),2009。
(16)salomon-ferrer,r.;
(17)case,d.s.;cheatham,iii,c.d.a.t.e.;darden,t.a.;duke,r.e.;giese,t.j.;gohlke,h.;goetz,a.w.;greene,d.homeyer,n.;izadi,s.;kovalenko,a.;lee,t.s.;legrand,s.;li,p.;lin,c.;liu,j.;luchko,t.;luo,r.;mermelstein,d.;merz,k.m.;monard,g.;nguyen,h.;omelyan,i.;onufriev,a.;pan,f.;qi,r.;roe,d.r.;roitberg,a.;sagui,c.;simmerling,c.l.;botello-smith,w.m.;swails,j.;walker,r.c.;wang,j.;wolf,r.m.;wu,x.;xiao,l.;yorkd.m.;kollman,p.a.2017,amber2017,加利福尼亚大学旧金山分校(universityofcalifornia,sanfrancisco)。
(18)wang,j.;wolf,r.m.;caldwell,j.w.;kollman,p.a.;case,d.a.《计算化学杂志》2004,25,1157-1174。
(19)bayly,c.i.;cieplak,p.;cornell,w.;kollman,p.a.《物理化学期刊》1993,97,10269-10280。
(20)besler,b.h.;merz,k.m.;kollman,p.a.《计算化学杂志》1990,11,431-439。
(21)singh,u.c.;kollman,p.a.《计算化学杂志》1984,5,129-145。
(22)jorgensen,w.l.;chandrasekhar,j.;madura,j.d.;impey,r.w.;klein,m.l.《化学物理学报(j.chem.phys.)》1983,79,926-935。
(23)wang,j.,cieplak,p.;kollman,p.a.《计算化学杂志》2000,21,1049-1074。
(24)darden,t.;york,d.;pedersen,l.《化学物理学报》1993,98,10089-10092。
(25)sondergaard,c.r.;olsson,m.h.m.;rostkowski,m.;jensen.,j.h.《化学理论与计算》2011,7(7)2284-2295。
(26)olsson,m.h.m.;sondergaard,c.r.;rostkowski,m.;jensen,j.h.“propka3:在经验性pka预测中对内部残留物和表面残留物的一致处理(propka3:consistenttreatmentofinternalandsurfaceresiduesinempiricalpkapredictions)。”《化学理论与计算》2011,7(2),525-537)。
实例2
mala的生化活性
通过ni-亲和色谱法和凝胶过滤纯化mala为体外测定提供纯蛋白,并且发现mala催化天然前体前畸枝霉酰胺(化合物2)的迭代氯化和溴化。在用一氯化化合物3和4进行的反应中,mala还能够使这些化合物氯化和溴化以产生化合物1、7、8和9,其中后三种是新颖的吲哚生物碱。通过与从金色畸枝霉分离出的标准品进行共洗脱来确认mala的氯化反应(图4)。1h-nmr分析确认吲哚环上的卤化位点和所有化合物的高分辨率质谱数据与卤化产物的预期质量和同位素峰模式一致。
卤化反应的转化百分比由分离的产率决定。化合物2在5mg体外反应中的氯化产生化合物3、4和1的转化率分别为34%、26%和24%。化合物2在4mg反应中的溴化产生按分离产率计23%的化合物5和18%的化合物6,但基于标准品曲线计算的转化率显示5与6对c9选择性之比为8:1。与先前报告相比,用于通过hplc使这些单卤化中间体分离的方法已得到很好的解决,因此向这些溴化吲哚生物碱的文献大大添加了单独分子的nmr数据。mala还被用作用于产生两种新颖的畸枝霉酰胺类似物,即化合物7和8的生物催化剂(图4和5)。化合物3在2mg反应中的溴化产生24%化合物7和15%化合物1作为副产物。化合物4在2.3mg反应中的溴化产生78%的化合物8。通过广泛的1d和2dnmr分析来确认化合物7、8和9的结构分配。使用关键ghmbcad相关性(图5)确认结构,其中观察到氯化碳与溴化碳相反的显著低场移动。吲哚环上卤素的位置通过在每个光谱的1h-nmr芳香族区中观察到的两个单峰确认。此外,化合物5和6还可以被氯化以分别产生化合物8和7。
实例3
mala的动力学表征
测定了mala的天然氯化反应的米-曼氏动力学参数。所述参数揭示了对于初始氯化反应和第二次氯化反应两者,酶具有类似的kcat和km值。化合物2与化合物3、化合物2与化合物4、化合物3与化合物1和化合物4与化合物1的kcat分别为0.08分钟-1、0.09分钟-1、0.12分钟-1和0.12分钟-1,这些值与fdhprna(0.10分钟-1)的那些值2相当。化合物2与化合物3、化合物2与化合物4、化合物3与化合物1和化合物4与化合物1的km值为7.0μm、7.5μm、4.4μm和4.0μm。计算了四个反应中的每个反应的催化效率,得出kcat/km值分别为11.5分钟-1毫米-1、12.0分钟-1毫米-1、27.3分钟-1毫米-1和29.7分钟-1毫米-1。与真核生物fdhrdc2的催化效率相比,这些催化效率相当高,初始氯化反应的催化效率为2.93分钟-1毫米-1,并且第二次氯化反应的催化效率为0.11分钟-1毫米-14。
实例4
mala'的底物复合物的结构表征
为了进一步阐明mala/mala'反应性在两个位点处的独特功能,测定了与前畸枝霉酰胺(化合物2)、畸枝霉酰胺b(化合物3)和异畸枝霉酰胺b(化合物4)复合的mala'的共晶体结构。mala和mala'的同一性为99%,仅两个氨基酸位置(mala中的leu276和arg428;mala'中的pro276和pro428)处不同并且具有相当的催化活性,但只有mala'易于结晶。为了验证mala'是mala的可行取代,对两者的活性进行比较,并且确定在测试的反应条件下mala'与mala基本上相同。分别在
trp263和trp265形成提出用于帮助辅因子结合的表征性黄素结合基序(图7)。与野生型mala相比,虽然malaw265a的活性急剧下降,但malaw263a的产物形成下降幅度较小(化合物4的产量为60%,化合物3的产量为50%,化合物1的产量为5%)。用组氨酸取代结合底物(phe489)的关键残基,以确定其在mala的活性中的重要性,并且发现活性也有所降低。phe489与苯丙氨酸类似,苯丙氨酸的诱变改变了fdhprna33中的位点选择性。
为了探讨mala的机制,glu494被包含丙氨酸、谷氨酰胺和天冬氨酸的各种其它残基取代。当e494a和e494q使mala灭活时,e494d保持少量活性。glu494与吲哚氮的质子形成氢键,从而促进底物的结合。天冬氨酸在此位置处的取代使底物远离催化赖氨酸移位,从而降低了活性(图8和10)。
探讨mala中的位点选择性机制的初始努力包含用丙氨酸、苯丙氨酸和其它氨基酸取代his253。malah253a对产生c9氯化化合物3具有选择性,而malah253f表现出对产生c8氯化化合物4具有选择性。与化合物2和3复合的mala'h253a的共晶体结构揭示了蛋白质中没有导致观察到的位点选择性的明显变化。另一方面,与化合物2复合的mala'h253f的共晶体结构揭示了s129(底物的吲哚环附近的残基)移位(图11)。当s129被丙氨酸取代时,malah253f的c8选择性被取消,从而得出ser129参与位置253处的phe取代诱导的选择性的结论(图8)。有趣的是,malah253a突变体和malah253f突变体未表现出对溴化反应的相同的位点选择性概况。相反,发现了与野生型mala相同的产物概况(参见图20)。
mala'的结构还揭示了与c末端附近的四个半胱氨酸残基(cys597、cys600、cys613、cys616)配位的独特的锌位点。zn2+离子是使用利用在包围锌k-边缘的x射线能量(9.6586kev)处记录衍射数据的反常散射实验鉴别的。仅使用来自边缘上方能量的数据在地图中显示异常差异密度。制备了双突变体malac613s/c616s,并且蛋白不可溶,因此在不存在zn2+的情况下未执行生化活性测定。
实例5
分子动力学模拟和qm模型
进行分子动力学(md)模拟,以从呈载脂蛋白和底物结合形式的mala'晶体结构开始获得对蛋白质的结构和活性的另外的见解。在后一种情况下,lys108氯胺中间体被认为与下文讨论的机制成一体。
对md轨迹的分析揭示了zn2+抗衡离子在蛋白质结构中发挥的结构作用。与非常柔性的相邻区(621-646)相比,zn2+结合区(597-616)内的残基表现出较低的均方根起伏(rmsf)。这些模拟表明,柔性区充当底物通道盖,其具有,这是在载脂蛋白结合状态和底物结合状态两者下在500纳秒md模拟期间被探索的两个主要的打开/关闭构象。
根据载脂蛋白状态轨迹,估计lys108和glu494侧链的pka。glu494的pka相对较高,为约6.0-7.5,而lys108的pka估计为7.2-8.3。
对底物与酶活性位点之间可能的极性相互作用的分析表明,尽管gly131和ala132的主链羰基可能潜在地与底物酰胺氮相互作用,但这些相互作用不如glu494–h(n-吲哚)氢键重要。后者对应于底物与蛋白质活性位点残基之间的主要且较强的相互作用,并且在整个md轨迹模拟期间都观察到所述相互作用利用所有结合底物(化合物2、3和4)。因此,glu494的碱性可以增强羧基侧链与底物的吲哚环之间的氢结合,从而使其与催化的lys108残基有效地相互作用。
包含提出的活性氯胺cl-lys108物种的md模拟表明,当底物2(化合物2)结合到活性位点中且通道盖关闭时,cl原子的位置非常靠近底物的c8/c9位置,这部分是因为glu494残基对底物的定位(参见图12a)。当盖处于其打开构象时,底物结合会发生轻微移位,但是仍与glu494进行h键结合,但随后cl-lys108侧链构象发生变化,以使cl原子更靠近fad辅因子。这表明底物结合的打开/关闭状态之间的蛋白质构象变化还控制了cl-lys108活性物种的定位,这可以探索两个主要构象,以将cl原子从黄素辅因子带到底物。此观察结果支持cl-lys108是实际的氯化物种的期望。当cl-lys108处于这种催化感受态时,针对c8位置和c9位置两者测量的距离(cl-c)和角度(cl-c-h)非常类似,这表明cl-lys108被预先组织为使两个位置氯化。
md模拟还揭示了cl-lys108与相邻asp109残基的主链羰基之间的关键相互作用,从而有效地将cl-lys108朝催化感受态排列定位(图12a)。当存在这种h键时,只能将来自lys108氯胺活性物种的cl原子置于c8/c9位置附近。通过诱变实验进一步探索了asp109的基本作用,并且d109a突变体的活性非常低(图8)。对此特定突变体的md模拟显示,ala109主链羰基指向的位置与原始asp109中的位置不同,从而消除了关键的氢键相互作用。这是由于氨基酸侧链的不同的构象所致,所述构象指向蛋白质腔外并且暴露于asp109中的溶剂中,而在ala109中则被置换。实际上,在d109a突变体的500纳秒模拟中,cl-lys108从未探索任何cl原子接近c8/c9位置以进行氯化的构象。
对具有结合底物2(化合物2)的mala'cl-lys108进行的md模拟突显了ser129侧链相对于化合物2中的h-c8的排列。当底物处于亲电芳香族取代的适当朝向时,ser129oγ与h-c8之间的距离特别短(介于
基于酶活性位点的实验证据和计算模型,研发了参与形成lys108氯胺中间体活性物种的mala卤化酶的机制(图12,方案2),然后所述物种与c8或c9相互作用以进行亲电芳香族取代(eas),在最终去质子步骤(方案2)之前产生韦兰德中间体(w)。这种去质子作用可以通过水分子充当碱来实现,或者在c8的情况下,通过良好定位的ser129来实现。为了深入了解所提出的反应机制,使用三种不同的计算模型进行密度泛函理论(dft)计算(有关计算细节,参见实例1)。第一模型仅将吲哚环和质子化的甲基氯胺视为活性物种(1a);第二模型靠近h-c8或h-c9位置添加水分子(1b);并且第三模型在h-c8位附近包含甲醇分子以模仿ser129(1c)。计算表明,初始氯化是反应的固有限速步骤,而去质子化步骤的速度稍快。与c8/c9质子相互作用的水分子或甲醇会加速氯化步骤,因为氢结合会增强这些碳的亲核性。通过配位的h2o(ts1b-c8)使计算出的c8氯化(ts1a-c8)的反应势垒从6.4kcal/mol降低到5.5kcal/mol,并且当甲醇与h-c8(ts1c-c8)配位时,反应势垒进一步降低到3.5kcal/mol。另一方面,通过h-c9(ts1b-c9)处的配位的h2o分子,计算出的c9氯化(ts1a-c9)的势垒从7.0kcal/mol降低到5.2kcal/mol,并且当甲醇与h-c8(ts1c-c9)相互作用时,势垒降到4.9kcal/mol,如图12所示。这突显了ser129在引导在形成c8氯化产物方面的选择性的作用。
wa-c9韦兰德中间体的稳定性比wa-c8的稳定性高1.3kcal/mol,但在考虑配位水分子(wb-c8和wb-c9)时,这些中间体变成几乎等能量。非极性环境有利于c9氯化产物3(化合物3)的形成。最终,一旦形成了韦兰德中间体,就会迅速发生通过去质子化的重新芳构化。当以水分子充当碱基时,对于c8(ts2b-c8)和c9(ts2b-c9),两个位置的计算出的去质子势垒分别为4.1kcal/mol和3.5kcal/mol,并且当甲醇充当碱基(ts2c-c8)时,c8的去质子势垒为0.6kcal/mol。
反应物复合物和过渡态的dft优化结构非常类似。在md模拟中发现了cl-lys108在c8和c9附近的催化感受态排列(如图12a、12c所呈现)。结合qm模型、md模拟以及cl-lys108对先前描述的底物的预组织,预计反应机制涉及cl-lys108中间体,这是mala黄素依赖性卤化酶的最合理的机制。
实例6
开发mala卤化酶作为后期c-h官能化的生物催化剂
畸枝霉酰胺是具有钙调蛋白拮抗剂的生物活性的复合的六环真菌吲哚生物碱,并且吲哚环的卤化对分子的效力具有显著贡献。mala已经被表征为迭代的后期卤化酶,所述卤化酶提供卤素部分以产生溴化和氯化的畸枝霉酰胺类似物。对卤化机制的实验研究已经鉴别活性位点内的丝氨酸残基对卤化的催化至关重要。此知识库已被用于工程化一系列mala变体,以在天然底物前畸枝霉酰胺上进行选择性卤化。对突变体的结构分析和计算分析有助于确定用于调节氯化反应的选择性的机制。mala催化反应的底物范围已通过筛选1,000种计算预测的底物进行了分析。本文所公开的实验工作已导致鉴别mala变体,即c9选择性突变体malag131s和s129a/i493s以及c8选择性突变体malas129a/p85s。参见图15。与变体相比,野生型酶的晶体结构有助于可视化这些突变如何改变结合袋并且提供对容纳非天然底物的见解。
参考文献
(1)weichold,v.;milbredt,d.;vanpée,k.-h.《应用化学国际版(angew.chem.int.ed.)》2016,55,2-18。
(2)dong,c.;flecks,s.;unversucht,s.;haupt,c.;van-pée,k-h.;naismith,j.h.《科学(science)》2005,309,2216-2219。
(3)yeh,e.;blasiak,l.c.;koglin,a.;drennan,c.l.;walsh,c.t.《生物化学》2007,46(5),1284-1292。
(4)zeng,j.;zhan,j.《化学生物化学(chembiochem.)》2010,11,2119-2123。
(5)ferrara,m.;perrone,g.;gambacorta,l.;epifani,f.;solfrizzo,m.;gallo,a.《应用及环境微生物学(appl.environ.microbiol.)》2016,82(18),5631-5641。
(6)neumann,c.s.;walsh,c.t.;kay,r.r.《美国国家科学院院刊(proc.natl.acad.sci.u.s.a.)》2010,107(13),5798-803。
(7)nielsen,m.t.;nielsen,j.b.;anyaogu,d.c.;holm,d.k.;nielsen,k.f.;larsen,t.o.;mortensen,u.h.《公共科学图书馆:综合(plosone)》2013,8(8),1-10。
(8)chankhamjon,p.;boettger-schmidt,d.;scherlach,k.;urbansky,b.;lackner,g.;kalb,d.;dahse,h.-m.;hoffmeister,d.;hertweck,c.《应用化学国际版》2014,53,13409-13413。
(9)sato,m.;winter,j.m.;kishimoto,s.;noguchi,h.;tang,y.;watanabe,k.《有机化学通讯(org.lett.)》2016,18,1446-1449。
(10)chankhamjon,p.;tsunematsu,y.;ishida-ito,m.;sasa,y.;meyer,f.;boettger-schmidt,d.;urbansky,b.;menzel,k.-d.;scherlach,k.;watanabe,k.;hertweck,c.《应用化学国际版》2016,55,11955-11959。
(11)cacho,r.a.;chooi,y.-h.;zhou,h.;tang,y.《acs化学生物学(acschem.biol.)》2013,8,2322-2330。
(12)menon,b.r.k.;brandenburger,e.;sharif,h.h.;klemstein,u.;shepherd,s.a.;greaney,m.f.;micklefield,j.《应用化学国际版》2017,10.1002/anie.201706342。
(13)yeh,e.;garneau,s.;walsh,c.t.《美国国家科学院院刊》2005,102(11),3960-3965。
(14)seibold,c.;schnerr,h.;rumpf,j.;kunzendorf,a.;hatscher,c.;wage,t.;ernyei,a.j.;dong,c.;naismith,j.h.;vanpée,k.-h.《生物催化生物转化(biocatal.biotransform.)》2006,24(6),401-408。
(15)zehner,s.;kotzsch,a.;bister,b.;süssmuth,r.d.;méndez,c.;salas,j.a.;vanpée,k-h.《化学生物学(chem.biol.)》2005,12,445-452。
(16)dorrestein,p.c.;yeh,e.;garneau-tsodikova,s.;kelleher,n.l.;walsh,c.t.《美国国家科学院院刊》2005,102(39),13843-13848。
(17)elgamal,a.;agarwal,v.;diethelm,s.;rahman,i.;schorn,m.a.;sneed,j.m.;louie,g.v.;whalen,k.e.;mincer,t.j.;noel,j.p.;paul,v.j.;moore,b.s.《美国国家科学院院刊》2016,113(14),3797-3802。
(18)martínez-luis,s.;rodríguez,r.;acevedo,l.;gonzález,m.c.;lira-rocha,a.;mata,r.《四面体(tetrahedron)》2006,62,1817-1822。
(19)watts,k.r.;loveridge,s.t.;tenney,k.;media,j.;valeriote,f.a.;crews,p.《有机化学期刊(j.org.chem.)》2011,76(15),6201-6208。
(20)figueroa,m.;gonzález-andrade,m.;sosa-peinado,a.;madariaga-mazón,a.;delrío-portilla,f.;delcarmengonzález,m.;mata,r.《酶抑制和药物化学杂志(j.enzymeinhib,med.chem.)》2011,26(3):378-385。
(21)madariaga-mazón,a.;hernández-abreu,o.;estrada-soto,s.;mata,r.《药学和药理学杂志(j.pharm.pharmacol.)》2015,67(4),551-558。
(22)klas,k.;tsukamoto,s.;sherman,d.h.;williams,r.m.;《有机化学期刊》2015,80,11672-11685。
(23)stocking,e.m.;williams,r.m.;《应用化学国际版》2003,42,3078-3115。
(24)finefield,j.m.;frisvad,j.c.;sherman,d.h.;williams,r.m.《天然产物杂志(j.nat.prod.)》2012,75,812-833。
(25)ding,y.;greshock,t.j.;miller,k.a.;sherman,d.h.;williams,r.m.《有机化学通讯》2008,10(21),4863-4866。
(26)li,s.;krithika,s.;tran,h.;yu,f.;finefield,j.m.;sunderhaus,j.d.;mcafoos,t.j.;tsukamoto,s.;williams,r.m.;sherman,d.h.《药物化学通信》2012,3,987-996。
(27)zeng,j.;zhan,j.《生物技术快报(biotechnol.lett.)》2011,33,1607-1613。
(28)buedenbender,s.;rachid,s.;müller,r.;schulz,g.e.《分子生物学杂志(j.mol.biol.)》2009,385(2),520-530。
(29)podzelinska,k.;latimer,r.;bhattacharya,a.;vining,l.c.;zechel,d.l.;jia,z.《分子生物学杂志》2010,397,316-331。
(30)hillwig,m.l.;liu,x.《自然化学生物学(nat.chem.biol.)》2014,10,921-923。
(31)gutekunst,w.r.;baran,p.s.《化学会评论(chem.soc.rev.)》2011,40,1976-1991。
(32)chung,w.;vanderwal,c.d.《应用化学国际版》2016,55,4396-4434。
(33)lang,a.;polnick,s.;nicke,t.;william,p.;patallo,e.p.;naismith,j.h.;vanpée,k.-h.《应用化学国际版》2011,50,2951-2953。
(34)glenn,w.s.;nims,e.;o'connor,s.e.《美国化学会志(j.am.chem.soc.)》2011,133,19346-19349。
(35)shepherd,s.a.;karthikeyan,c.;latham,j.;struck,a.-w.;thompson,m.l.;menon,b.r.k.;styles,m.q.;levy,c.;leys,d.;micklefield,《化学科学杂志(j.chem.sci.)》2015,6,3454-3460。
(36)shepherd,s.a.;menon,b.r.k.;fisk,h.;struck,a.-w.;levy,c.;leys,d.;micklefield,j.《化学生物化学》2016,17,821-824。
(37)payne,j.t.;andorfer,m.c.;lewis,j.c.《应用化学国际版》2013,52,5271-5274。
(38)payne,j.t.;poor,c.b.;lewis,j.c.《应用化学国际版》2015,54,4226-4230。
(39)andorfer,m.c.;park,h.j.;vergara-coll,j.;lewis,j.c.《化学科学(chem.sci.)》2016,7,3720。
(40)megalign
如从上下文中显而易见的,本申请中提及的所有出版物和专利均通过引用以其整体或相关部分并入本文。在不背离本公开的范围和精神的前提下,对所公开的主题作出的各种修改和改变对本领域的技术人员而言是显而易见的。尽管已结合特定的优选实施例描述了本公开,但应当理解,不能将所要求保护的本发明不适当地限定为此类特定实施例。实际上,用于制作或使用所公开的主题所描述的方式的各种修改旨在处于下列权利要求的范围之内,其中所述修改对于一个或多个相关领域的技术人员是显而易见的。
- 还没有人留言评论。精彩留言会获得点赞!