设计的、有效的和广泛特异性的有机磷酸酯水解酶的制作方法
相关申请
本申请要求2018年8月14日提交的以色列专利申请号261157的优先权权益,其内容以其整体通过引用并入本文。
序列表声明
与本申请的申请同时提交的,题为78359sequencelisting.txt、在2019年8月14日创建、包含188,416字节的ascii文件通过引用并入本文。
发明领域和背景
在其一些实施方案中,本发明涉及酶学,且更具体地但非排他地涉及通过指定的计算方法设计以表现出对广泛范围的有机磷酸酯和化学战争神经试剂的催化活性的磷酸三酯酶变体。
目前,化学战争神经试剂(cwna)中毒的预防和中毒后治疗均基于选择用于抵消由胆碱能神经元中乙酰胆碱的积累引起的症状的药物。当前的解毒方案由以下组成:用吡斯的明预处理,以及涉及施用含有阿托品、肟再活化剂和抗惊厥剂、诸如地西泮的鸡尾酒的暴露后疗法。针对cwna毒性的多药方法已被许多国家采用,并已整合入其民用和军用医疗方案。然而,通常认识到这些药物方案受困于几个缺点,其需要新的治疗策略。优选的方法是在血液中的cwna有机会到达其生理靶标之前将其迅速解毒。实现该目标的一种方式是通过使用生物清除剂。然而,使用目前可用的最佳化学计量生物清除剂(人丁酰胆碱酯酶,hbche)需要施用数百毫克的蛋白,以赋予针对毒性剂量的cwna的保护。
通过使用催化生物清除剂快速降解循环中的中毒有机磷酸酯(op),可以实现更安全且更有效的治疗策略。磷酸三酯酶(pte)的混杂神经试剂水解活性使其成为神经试剂中毒的预防和暴露后治疗的主要候选药物。然而,在暴露于毒性剂量的神经试剂后使用低剂量的酶(≤50mg/70kg)进行有效的体内解毒需要将增加野生型pte对毒性神经试剂异构体的催化效率(kcat/km)。
先前公开了可以有效地水解v-型神经试剂的pte变体[cherney,i.等人,acschembiol,2013,8(11),pp.2394-2403]。这些变体之一(c23)的体内暴露后活性在用致死剂量的vx中毒的豚鼠中得到证实[worek,f.等人,toxicollett,2014,231(1),pp.45-54]。
关于pte变体的额外
背景技术:
包括美国专利号8,735,124,wo2016/092555,wo2018/087759以及roodveldt,c.和tawfik,d.s.,proteinengdessel.,2005,18(1),pp.51-8。
改变酶活性概况的突变对于适应生物体变化的需求(诸如代谢新的底物)至关重要。在基础研究、生物技术和生物医学中也非常期望此类突变,以实现有效和环境安全的解决方案,例如在有用分子的合成或有害分子的降解中。然而,大多数突变对蛋白活性和稳定性是有害的,限制了通过自然进化或蛋白工程改造改进的变体的出现。此外,由于突变上位性(mutationalepistasis),突变对活性的影响取决于先前是否获得其他突变。在极端情况下(称为标志上位性(signepistasis)),单独有害的两个突变当组合时增强活性,或反之亦然。在自然进化中,突变通常一次发生一个,且因此,突变的上位组合必须以特定顺序累积,因为所有中间体必须至少与其前身一样有活性,或者它们将通过选择清除。在改进的突变体中的标志上位性的高流行进一步降低获得有利组合的可能性。蛋白进化还受到稳定性-阈值效应的限制,由此增强活性的突变可能使蛋白不稳定,且因此仅累积直至其中不再耐受额外突变的阈值。为了克服稳定性-阈值效应,在活性位点口袋附近和远处区域中的稳定突变对于积累功能增强突变是必需的。
由于上位性和稳定性-阈值效应,酶活性显著增强的变体的进化需要不同类型的多重突变并影响蛋白的不同区域。例如,实验室进化实验可能包括十几轮遗传多样化和对改进突变体的选择,并且三个或更多个数量级的显著提高平均需要十个突变。这些突变大多数发生在催化口袋之外,并且可能仅通过增强对功能增强突变的耐受性而间接影响活性。另一个复杂因素是实验室进化实验是费力的,并且需要高通量或甚至超高通量筛选(每轮>106个变体)。然而,此类筛选仅适用于某些酶活性,并且通常采用合成模型底物。
原则上,计算蛋白设计策略可以绕过多轮实验优化的需求,因为它们不受突变轨迹的限制。蛋白设计的先前应用计算了有利的点突变体或聚焦用于实验筛选的文库,得到活性的有限增加,并且从头设计的酶表现出低催化效率。总体而言,计算酶设计仍然是一种专门技术,并且仍取决于实验室进化以达到与天然酶中所见的那些相当的效率。因此,在酶设计的基本原理的理解和控制中仍然存在显著差距。
关于蛋白变体的计算设计的额外背景技术包括美国专利申请公开号2017/0032079,国际专利申请号wo2017/017673,fleishman,s.l.等人,plosone,2011,6(6),和goldenzweig,a.等人molcell.,2016,63(2),pp.337-346。
发明概述
酶活性的显著提高需要在活性位点中的空间近端位置进行多重突变。然而,此类突变经常表现出对活性不可预测的上位(非累加)作用。在此,本发明提供了一种使用系统发生分析和rosetta设计计算在酶活性位点处设计多点突变的自动化方法,在本文中称为funclib。本文使用磷酸三酯酶证明funclib;pte的设计变体都有活性,并且大多数显示与野生型和与彼此显著不同的活性概况。仅具有3-6个活性位点突变的数十种设计对一定范围的替代底物(包括毒性有机磷酸酯神经试剂soman和环沙林的水解)表现出高10-4,000倍的效率。funclib也已作为网络服务器(www(dot)funclib(dot)weizmann(dot)ac(dot)il)实现;其规避了迭代的高通量筛选,并为设计高效和多样化的催化库打开了道路。
因此,根据本发明的一些实施方案的一个方面,提供了蛋白,其具有选自对作为原始蛋白的来自缺陷假单胞菌(pseudomonasdiminuta)的磷酸三酯酶(pte)提供并且在表a中列出的序列空间的至少2个氨基酸取代的任何组合的序列:
在一些实施方案中,所述蛋白是杂合蛋白,其中氨基酸取代的组合在除了原始蛋白以外的pte蛋白上执行。
在一些实施方案中,所述蛋白的特征在于选自下文所述的表a中呈现者的序列。
在一些实施方案中,所述蛋白的特征在于选自以下的序列:pte_28(seqidno:28)、pte_29(seqidno:29)、pte_56(seqidno:56)和pte_57(seqidno:57)。
根据本发明的一些实施方案的一个方面,提供了有机磷酸酯试剂的解毒和去污的方法,其通过如下实现:使怀疑被有机磷酸酯试剂污染的区域与根据本发明的一些实施方案在本文中提供的pte变体蛋白中的至少一种接触。
在一些实施方案中,所述区域选自地板、墙壁、建筑物或其部分、车辆、一片衣服、一片设备、植物、动物和无生命的物体。
在一些实施方案中,所述有机磷酸酯试剂选自g-型神经试剂、v-型神经试剂和gv-型神经试剂。
根据本发明的一些实施方案的一个方面,提供了生成酶变体(设计)的文库的方法,所述酶变体与原始酶相比具有多样的提高的催化活性,所述方法通过如下实现:
鉴定所述酶的活性位点的第一壳和第二壳中的一组可取代残基(可取代位置),以及这些壳中的一组固定残基(固定位置);
根据pssm评分方案使用计算软件重新排列所述可取代残基的突变,所述计算软件计算稳定性参数并根据其能量值对重新排列的突变体进行排序,由此获得酶变体的稳定性评分列表;
列举由前一步骤产生的酶变体;
在所述稳定性评分列表的顶部选择许多所得的变体(重新排列的突变体),与所述原始酶相比,所述变体在所述可取代残基中具有至少两个突变;和
克隆和表达该许多相对于所述原始酶具有顶级稳定性评分和至少两个突变的变体。
在一些实施方案中,生成酶变体的文库的方法进一步包括,在鉴定可取代和固定的残基之前,使用任何稳定性设计方法(诸如pross)和使用该变体作为原始酶提供野生型酶的稳定变体。
除非另有定义,否则本文使用的所有技术和/或科学术语具有与本发明所属领域普通技术人员通常理解相同的含义。尽管下文描述了示例性方法和/或材料,但与本文描述的那些类似或等效的方法和材料可以用于本发明的实施方案的实践或测试中。在冲突的情况下,以专利说明书(包括定义)为准。另外,材料、方法和实例仅是说明性的,并且不预期一定是限制性的。
本发明的实施方案的方法和/或系统的实现可以涉及人工、自动或其组合执行或完成所选任务。此外,根据本发明的方法和/或系统的实施方案的实际使用仪器和装备,几个所选任务可以使用操作系统通过硬件、通过软件或通过固件或通过其组合来实现。
例如,根据本发明的实施方案用于执行所选任务的硬件可以作为芯片或电路实现。作为软件,根据本发明的实施方案的所选任务可以使用任何合适的操作系统作为通过计算机执行的多个软件指令实现。在本发明的一个示例性实施方案中,根据如本文描述的方法和/或系统的示例性实施方案的一个或多个任务通过数据处理器,诸如用于执行多个指令的计算平台来执行。任选地,数据处理器包括用于储存指令和/或数据的易失性存储器和/或用于存储指令和/或数据的非易失性存储器,例如磁性硬盘和/或可移动介质。任选地,同样提供了网络连接。同样任选地提供了显示器和/或用户输入装置、诸如键盘或鼠标。
附图的几个视图的简述
本发明的一些实施方案在本文中参考附图仅作为实例进行描述。现在具体参考详细附图,要强调的是所示的细节作为实例且用于本发明的实施方案的说明性讨论的目的。在这方面,与附图结合的描述使得本发明的实施方案可以如何进行实践对于本领域技术人员显而易见。
在附图中:
图1a-d举例说明用于从细菌pte(pdb条目:1hzy)的结构以及稳定的变体或pte,dpte2(seqidno:1)的序列开始产生功能性磷酸三酯酶酶库的计算设计方法中的关键步骤,其中图1a呈现这样的步骤,其中选择活性位点位置用于设计,并且在每个位置,序列空间受到进化保守分析(pssm)和突变扫描计算(δδg)的约束,图1b呈现其中使用rosetta原子设计计算详尽列举多点突变体的步骤,图1c呈现其中根据能量对设计进行排序的步骤,且图1d呈现这样的步骤,其中将序列聚类以获得用于实验测试的多样化、低能量(即稳定和预先组织)设计的库,而设计位置在所有小图中始终一致地着色;
图2a-c呈现使用根据本发明的实施方案的方法funclib的一些结果,其中设计的磷酸三酯酶(pte)库表现出一定范围混杂活性的数量级的提高(图2b的x-轴中的数字和图2c中的y-轴中的数字代表变体编号(pte_x)和seqidno:x);
图3呈现的图显示,根据本发明的一些实施方案,本文提供的pte变体中的设计突变表现出标志-上位性关系,其中每个圆圈代表dpte2(seqidno:1)的突变体,每个圆圈的面积与所述变体在水解芳酯乙酸2-萘酯(2na)中的比活性成正比,且其中pross设计和稳定的序列dpte2(seqidno:1)(其被用作本文提供的方法中的起点)表现出低比活性,并且每种点突变体表现出提高的比活性,比活性在双重突变体中下降,并且四重突变体设计pte_6(seqidno:6)相对于所有单一或双重突变体实质上提高比活性;且
图4呈现作为根据本发明的一些实施方案在本文提供的pte变体中的选择性变化的基础的设计的活性位点口袋的立体化学特性的图示,其中pte_28(seqidno:28;在图4中表示为28)和pte_29(seqidno:29;在图4中表示为29)表现出比dpte2(seqidno:1;在图4中表示为1)更大的活性位点口袋以及针对大体积的v-和g-型神经试剂的高催化效率(从左上角起按顺时针顺序,分子渲染基于pdb条目:1hzy、6gbj、6gbk和6gbl;球体指示双金属中心的离子。
本发明的具体实施方案的描述
在其一些实施方案中,本发明涉及酶学,并且更具体地但非排他地涉及通过指定的计算方法设计以表现出对广泛范围的有机磷酸酯和化学战争神经试剂的催化活性的磷酸三酯酶变体。
在详细解释本发明的至少一个实施方案之前,应理解本发明在其应用中不一定限制于下述说明书中阐述和/或附图和/或实施例中举例说明的计算、计数和计算参数和/或实验室方法的值的细节。本发明能够具有其他实施方案或者以各种方式实践或实施。
在详细解释本发明的至少一个实施方案之前,应理解本发明在其应用中不一定限制于下述说明书中阐述或通过实施例例举的细节。本发明能够具有其他实施方案或者以各种方式实践或实施。
用于设计酶的功能多样化库的方法:
为了解决仍然困扰当代蛋白设计方法的空白,如上文引言部分中所讨论,本发明人已经开发了一种蛋白设计策略,其提供了在活性位点处具有稳定的相互作用残基网络的蛋白序列,并选择了适用于低通量筛选的多样设计的小集合。本文提供的这种设计范例和实践策略以及相应的计算工具和方法,通过设计相互作用的活性位点多点突变体的密集且预先组织的网络来解决上位性。任选地,蛋白设计策略可以进一步包括使用通过首先设计稳定的酶支架来解决稳定性-阈值效应的pross。该方法没有先验地靶向特定的底物,因为这要求酶过渡态复合物的精确模型,并且此类模型很少可获得,并且大多是近似的。反而,根据本发明的一些实施方案,本文提供的方法(设计策略)导致可以针对目标活性进行筛选的稳定且高效的蛋白(例如,酶、抗体等)的库。
如本文所呈现,为了证明性目的从示例性酶开始,本文提供的方法用于设计功能多样的库,其包含数十种在一定范围活性中表现出10-4,000倍提高的酶。本文呈现的策略的稳健性和有效性可以与先前提供的方法、实施的公开可得的蛋白-稳定平台“pross”组合(参见,美国专利申请公开号2017/0032079和wo2017/017673,其各自通过引用并入本文,如同在本文完整阐述一样;以及例如www(dot)pross(dot)weizmann(dot)ac(dot)il/)。与其一起提供并称为“funclib”或“ablift”的方法也已作为自动化的网络可访问服务器实现。
pross和本文提供并在funclib和ablift中实现的方法之间的主要差异在于,pross在活性/结合位点之外设计蛋白,而funclib和ablift设计活性/结合位点,因为pross的目标是稳定蛋白,而不改变其结构相关的活性。这种区别是至关重要的:由于任何蛋白中存在许多位置用于设计稳定的变体(>90%的蛋白与功能不直接相关),因此pross使用组合设计算法来仅寻找最安全的突变组合,所述组合设计算法假设骨架保持固定,并导致突变的组合,其具有对稳定性的主要累加影响。相比之下,funclib/ablift在其中位置高度相互依赖的蛋白系统区域(活性/结合位点)中起作用。在此类结构区域中,存在较少允许的突变(<=10%的蛋白,并且由于功能限制,保守性非常高),并且几乎所有位置都彼此依赖,因此几乎没有“安全”的突变组合,其中每个突变以累加的方式影响活性;它们都是潜在有害的,而且实验确实显示,这些区域对突变难以置信地敏感,更不用说多点突变了。因此,在本文提供且作为示例性程序funclib和ablift实施的方法中,首先使用比pross更宽松的设置(能量稳定性阈值)鉴定耐受的序列空间,以便甚至在保守位置中也能够进行突变,且其次列举所有可能的组合,将其保持在可管理的数量以实现有效的计算。在通过本文提供的方法(funclib/ablift)生成的多点突变体的每个实例中,允许骨架改变构象,由此允许突变,包括被认为对计算设计非常困难的小至大的突变和甚至小至大的突变的组合。然后,根据能量对所有列举的多点突变体进行排序,以确保仅选择稳定的、预先组织的突变网络。本发明的发明人已经令人惊讶地注意到,经常存在数百种或甚至数千种具有比野生型或原始/起始序列更低的能量(更稳定)的序列,这通过应用直接的组合设计模拟或在pross结果中从未见到。因此,本文提供的方法基于对序列空间的严格取样,对蛋白的刚性或突变对功能或稳定性的累加贡献的假设较少。
尽管funclib和ablift共享许多计算组分,但本文提供的计算蛋白设计方法的两种实施方式之间的主要差异在于funclib主要应用于酶活性位点,所述酶活性位点是溶剂暴露的,且因此可能仍然对突变耐受,而将ablift应用于两条蛋白链之间的界面(例如,抗体中的轻/重链界面)。该链界面区域与蛋白核心一样紧密地堆积,且因此潜在地对突变的耐受性较低。在本文中注意到,先前提供的方法pross通常无法在此类区域中找到突变,并且ablift被指定为容易地找到数百种具有提高的能量(稳定性和预先组织性)的多点组合。
因此,本文提供的方法(funclib/ablift)处理的问题是如何在高度保守的区域中的相互依赖的位置间找到有利的多点突变体-pross明确试图避免这种结果,其他计算设计一般通常失败,并且实验体外进化策略经常需要多次迭代逐步筛选才能实现。
因此,根据本发明的一些实施方案的一个方面,提供了一种用于从模板/原始蛋白(原始多肽链)、例如酶开始计算设计蛋白(多肽)的文库的方法,其中与模板/原始蛋白相比,该文库的成员表现出一定范围的活性和功能的10-4,000倍提高。在一些实施方案中,所述蛋白是在底物/产物/速率方面具有已知活性的酶,并且根据本发明的实施方案生成的文库包括具有提高的已知活性和/或新活性中的任一种或两者的酶。注意,在本发明的背景下,新活性可以被视为已知为低或基本为无效的活性,因此下面的描述同时解决新的和提高的活性,因为提高可以从基本上没有活性开始直至增强的活性,无论已知活性如何。
在参数值和rosetta能量单位的方面,与pross中使用的能量稳定性阈值(其包括pssm评分≥0和δδg评分≤-0.45、-0.9、-2.0、-3.0或-4.0)相比,在funclib/ablift中使用的更松散的能量稳定性阈值包括pssm评分≥-2或-1和δδg评分≤+1、+2、+3、+4、+5或+6。
为了证明该方法,选择具有公开可得的晶体结构的酶,来自缺陷假单胞菌(pseudomonasdiminuta)的含锌磷酸三酯酶(pte)(pdb条目1hzy)。本文呈现的方法有效地用于提供修饰的多肽链,从原始多肽链开始,诸如在相应的野生型蛋白或先前工程改造/设计的变体中所发现,其中将原始多肽链中的几个氨基酸残基取代,使得经表达以具有修饰的多肽链的蛋白(变体蛋白)表现出与野生型蛋白相比提高的就特定底物而言的催化活性以及结构稳定性。如本文所用的术语“变体”是指通过采用本文呈现的方法获得的设计蛋白。本文以及全文中,术语“氨基酸序列”和/或“多肽链”还用作对具有该氨基酸序列和/或该多肽链的蛋白的提及;因此术语“原始氨基酸序列”和/或“原始多肽链”等于或涉及术语“原始蛋白”和“野生型蛋白”,并且术语“修饰的氨基酸序列”和/或“修饰的多肽链”和/或“设计的多肽”等于或涉及术语“设计的蛋白”和“变体”。
在一些实施方案中,原始多肽链,或原始蛋白,是天然存在的(野生型;wt)或人工的(人造的非天然存在的)或设计的多肽链,即计算方法(诸如pross)的产物。
在本发明一些实施方案的背景下,术语“设计的”及其任何语法变化,是指非天然存在的序列或蛋白。
在本发明的一些实施方案的背景下,当提及具有特定序列的特定蛋白时,术语“序列”与术语“蛋白”可互换地使用。
根据本发明的一些实施方案的一个方面,提供了从原始多肽链开始计算设计修饰的多肽链的方法。
图1a-d是根据本发明的一些实施方案,用于执行从原始多肽链开始计算设计修饰的多肽链的方法的示例性算法的示意图说明。
方法要求和输入准备:
用于执行用于为了活性多样性而设计修饰的多肽链的方法的基本要求包括:
以下信息的可用性:关于原始多肽链的结构信息,诸如获得自实验确定的原始多肽链的晶体结构或具有至少30-60%氨基酸序列同一性的其亲近的同源物的晶体结构,或基于其亲近的同源物的实验确定的结构计算得到的结构信息;
实验性突变分析(点突变、突变组合或深度突变扫描)的任选可用性;和
从几种合格的同源蛋白的得到的序列数据的可用性,而对于合格的同源序列的标准在下文描述(图1a)。在同源蛋白的低利用率的一些情况下,所述方法利用一种独特的方法来选择合格的同源序列,如下所述。
在本发明的实施方案的背景下,如在本领域中,术语“%氨基酸序列同一性”或简写“%同一性”在本文中用于描述在比对中两个氨基酸序列在相同位置具有相同残基的程度。应指出的是术语“%同一性”也用在核苷酸序列的背景下。
本文中注意到,通常,本文呈现的方法(例如,funclib)不需要过渡态或其复杂结构的结构模型。相反,它计算活性位点口袋处相互作用残基的多样而稳定的网络,由此为不需要先验地定义的替代底物/配体编码不同的立体化学互补性。因此,期望该方法提供形成功能库的设计,从所述功能库可以分离有效翻转各种靶标底物的单独设计。相反,在靶向特定底物的应用中,可以通过在底物或过渡态模型存在的情况下设计酶来进一步限制序列空间,并且可以在本文呈现的网络服务器中启用该选项。
结构数据准备:
根据本发明的一些实施方案,结构信息为原始多肽链的原子坐标的集合。该原子坐标集合在本文中称作“模板结构”,其在下文所讨论的方法中使用。在一些实施方案中,模板结构为原始多肽链的晶体结构,并且在一些实施方案中模板结构为基于原始多肽链的亲近的同源物(超过30-60%同一性)的晶体结构计算生成的结构,其中原始多肽链的氨基酸序列已经穿引在其上并进行加权拟合以提供其能量最小化,这些在下文中讨论。
在其中目标蛋白为寡聚体(具有几条多肽链)的情况下,目标链或待修饰的原始多肽链在模板结构中定义。在杂寡聚体的情况下,需要选择将经历序列设计程序的链或使两条链进行同时设计。对于同寡聚体,选择具有更多或更好质量的结构数据的原始多肽链是有利的。例如,在一些同寡聚体中,结合离子可能在一些链中在晶体结构中可辨认,但在其他中不足以此。另外,定义与功能和活性相关的关键残基是有利的,如下文中所讨论的。
结构精修:
根据一些实施方案,在用于本文呈现的方法之前,模板结构任选地进行全局能量最小化,由其加权拟合提供,如下文所讨论的。
根据本发明的一些实施方案,模板结构任选地在使用其坐标之前通过能量最小化精修,同时固定关键残基的构象,如下文所定义的。结构精修为计算化学中的常规程序,并且通常涉及基于自由能最小化的加权拟合,进行规则,诸如谐波抑制(harmonicrestraint)。
根据本发明的任何实施方案的一些实施方案,术语“加权拟合”是指一个或多个计算结构精修程序或操作,旨在通过基于涉及,例如,精修结构的序列同源性评分、骨架二面角和/或原子位置(变量)的预定的权重、抑制和约束(常量)通过使多项式函数最小化来优化几何学、空间和/或能量标准。根据一些实施方案,加权拟合程序包括调整键长和键角、骨架二面(ramachandran)角、氨基酸侧链堆积(旋转异构体)和氨基酸的迭代取代中的一种或多种,而术语“调整键长和键角”、“调整骨架二面角”、“氨基酸侧链堆积”和“改变氨基酸序列”在本文中也用于指,尤其,广泛用在计算化学和生物学领域中的众所周知的优化程序和操作。根据本发明的一些实施方案,示例性的能量最小化程序为循环坐标下降(ccd),其可以用用于大分子建模的rosetta™软件套装中的默认的所有原子能量函数执行。对于一般优化程序的综述,参见例如,christodoulosa.floudas和panosm.pardalos的“encyclopediaofoptimization”,springerpub.,2008。
根据本发明的一些实施方案,用于执行本文呈现的方法的合适的计算平台为rosetta™软件套装平台,从美国华盛顿大学baker实验室的“rosetta@home”公开可用。简言之,rosetta™是用于理解蛋白结构、蛋白设计、蛋白对接、蛋白-dna和蛋白-蛋白相互作用的分子建模软件包。rosetta软件包含多个功能模块,包括rosettaabinitio、rosettadesign、rosettadock、rosettaantibody、rosettafragments、rosettanmr、rosettadna、rosettarna、rosettaligand、rosettasymmetry等。
根据一些实施方案,加权拟合在一组抑制、约束和权重(称为规则)下实现。例如,当精修具有第一个构象的任何给定的多肽区段的骨架原子位置和二面角时,为了向不同的第二个构象驱动、同时试图尽可能保存第二个构象中观察到的二面角,计算程序将会使用偏倚,例如,cα位置的谐波抑制和偏倚自由背离第二个构象中观察到那些的骨架二面角的谐波抑制,因此允许每一结构决定簇发生最小构象变化,同时驱动整体骨架变为第二个构象。
在一些实施方案中,全局能量最小化是有利的,这是由于用于确定和精修模板结构源的能量函数和本文呈现的方法所使用的能量函数之间的差异。通过在骨架构象和在旋转异构体构象中通过最小化允许发生变化,全局能量最小化解除小的错配和小的立体冲突,由此使一些模板结构的总自由能降低显著的量。
在一些实施方案中,能量最小化可包括旋转异构体取样(重新堆积)、随后为侧链和骨架最小化的迭代。示例性的精修方案在korkegian,a.等人,science,2005中提供。在一些实施方案中,能量最小化可以包括蛋白的骨架中更实质性的能量最小化。
如本文所用,术语“旋转异构体取样”和“重新堆积”是指特定的加权拟合程序,其中将有利的侧链二面角取样,如rosetta软件包中所定义。重新堆积通常将较大的结构变化引入加权拟合的结构中(与标准二面角最小化相比),因为后者样品小的残基构象变化,而重新堆积可使侧链绕二面角旋转,使得其在蛋白结构中占据完全不同的空间。
在一些实施方案中,其中模板结构为同源蛋白的,首先使用充分建立的计算程序将查询序列穿引在蛋白的模板结构上。例如,当使用rosetta软件包时,根据本发明的一些实施方案,前两次迭代以“软”能量函数进行,其中原子半径定义为较小。较小半径值的使用减少强排斥力,导致较平稳的能量景观并允许跨越能量障碍。下一次迭代用标准rosetta能量函数进行。“坐标约束”项可加至标准能量函数以允许与原始cα坐标的实质性偏差。坐标约束项协调表现(hooke法则),具有范围在约0.05-0.4r.e.u(rosetta能量单位)之间的权重,取决于查询序列和模板结构的序列之间的同一性程度。在精修期间,关键残基仅进行小范围最小化但不进行旋转异构体取样。
序列数据准备:
一旦已经鉴定原始多肽链并且已经提供相应的模板结构,所述方法需要组装与原始多肽链的氨基酸序列相关的合格的同源氨基酸序列的数据库。原始多肽链的氨基酸序列可以,例如,从fasta文件提取,所述fasta文件对于蛋白通常从蛋白数据库(pdb)可得或另外提供。根据本发明的一些实施方案,对合格的同源序列的搜索在非冗余(nr)蛋白数据库中使用原始多肽链的序列作为搜索查询进行。这种nr-数据库通常包含手动和自动地注释的序列,且因此远大于仅包含手动注释的序列的数据库。
蛋白序列数据库的非限制性实例包括insdcembl-bank/ddbj/genbank核苷酸序列数据库、ensembl、flybase(对于昆虫家族果蝇科)、h-invitational数据库(h-inv)、国际蛋白索引(internationalproteinindex)(ipi)、蛋白信息资源(proteininformationresource)(pir-psd)、蛋白数据库(proteindatabank)(pdb)、蛋白研究基金会(proteinresearchfoundation)(prf)、refseq、酵母基因组数据库(saccharomycesgenomedatabase)(sgd)、拟南芥信息资源(thearabidopsisinformationresource)(tair)、trome、uniprotkb/swiss-prot、uniprotkb/swiss-prot蛋白同种型、uniprotkb/trembl、脊椎动物和基因组注释数据库(vertebrateandgenomeannotationdatabase)(vega)、wormbase、欧洲专利局(epo)、日本专利局(jpo)和美国专利局(uspto)。
在nr-数据库中搜索产生可变的结果,取决于搜索查询(原始多肽链的氨基酸序列)。对于缺乏序列数据的蛋白,结果可包括小于10个命中物。对于所有生命界所共有的蛋白,结果可包括数千个命中物。对于大多数蛋白,在nt-数据库中搜索后,数百至数千个命中物是预期的。在所有数据库中,包括nr-数据库,并且不论其名字为何,可能存在一些程度的冗余,并且命中物可能在同一序列的组中存在。冗余问题在序列数据编辑期间解决。
在本发明的一些实施方案中,将获得的序列数据任选地如下过滤和编辑:
(a)将冗余序列聚类(cluster)为单一的代表性序列。聚类用预定阈值进行。例如,阈值0.97意指将其自身中共享至少97%同一性的所有序列聚类为单一代表性序列,所述单一代表性序列是贡献于聚类的所有序列的平均值;
(b)排除比对长度小于搜索查询长度的预定阈值(例如,60%)的序列;和
(c)排除例如关于搜索查询表现出低于约28%-34%同一性截止值的序列,其遵循诸如别处提供的指南[rost,b.,proteineng,1999,12(2):85-94]。
最小的同一性参数的准确选择取决于序列数据的丰度。因此,根据本发明的一些实施方案,如果在严格的阈值下提供的序列命中物的数目为约50或更少,可使用不太严格的阈值(较低的%同一性)。同一性参数的阈值调谐(tuning)的作用在设计来自缺陷假单胞菌的磷酸三酯酶中得到证明,其中将阈值从30%同一性降至28%同一性,使合格的同源序列的数目从45增加至95。
在本发明的一些实施方案中,用于选择用于多序列比对的合格的同源序列的截止值为相对于原始多肽链大于20%、25%、30%、35%、40%或大于50%同一性。
应指出的是所述方法不限于任何特定的序列数据库、搜索方法、同一性确定算法和用于使同源序列有资格的任何标准集合。然而,通过使用所述方法获得的结果的质量在一定程度上取决于输入序列数据的质量。
一旦获得合格的同源序列的集合,生成多序列比对(msa)(图1a),这通常通过使用指定的多序列比对算法,诸如muscle中执行的算法[edgar,r.c.,nucleicacidsres,2004,32(5):1792–1797]。或者,basiclocalalignmentsearchtool(blast)可用于生成msa文件。
同源蛋白的利用率低的情况:
通常,将表现出低于20%的%同一性的序列添加至具有数十种较高%同一性的同源序列的msa中可能有助于比对的多样性;然而,添加此类低%同一性序列显著增加错误(假阳性)的风险,而并不一定大大提高多样性,因为这种多样性的大部分可能被已经是msa的部分的高同源性序列所覆盖。另一方面,当目标蛋白在序列数据库中代表不佳时,使用低%同一性同源物成为优势,而不是风险。
在一些情况下,在非冗余同源序列的数量的方面,目标蛋白在目前可用的蛋白序列数据库中代表不佳。例如,在序列同源性搜索仅发现与目标蛋白具有60%序列同一性的一个同源序列的情况下,这意味着该方法限于在60%的序列位置中的零个氨基酸取代,并且在剩余40%中,会难以鉴定具有多于几个氨基酸替代物的位置。
在此类情况下,本发明人已经设想了几种情形,其中标准序列同源性搜索方法可能导致同源序列空间内的低序列多样性(例如,小于50%、小于40%、小于30%、小于25%(“暮色区”)或相对于目标蛋白的氨基酸序列小于20%序列同一性。这种情形的一个实例是其中目标蛋白(靶标蛋白,在本文中也称为原始多肽链)的折叠是独特的或在系统发生上限于特定的属或门,或者蛋白功能已在近千年中出现,并且因此目标蛋白几乎没有同源物。本发明人预见到,在低序列多样性的此类或其他情况下,可以采取以下步骤来增加当前提供的方法使用的序列多样性,同时使引入无关序列的风险最小化。
用于处理此类情况的示例性子算法描述于美国专利申请公开号2017/0032079,其通过引用并入本文。该子算法背后的一般原理是,尽可能多地增加msa中的同源序列的数量,同时使包括不相关序列的风险最小化;例如,考虑到以下事实:目标蛋白的折叠是独特的和/或与通过测序工作询问的典型生物在系统发生上相距遥远。
步骤1:在任何给定序列数据库中通过使用专门检测远距同源物的算法(例如,csi-blast;参见,pmid:19234132,18004781)搜索低序列同一性同源序列(例如,小于50%,小于40%,小于30%,小于25%或小于20%序列同一性;优选小于30%同一性);
步骤2:使用聚类阈值90-100%对来自步骤1的结果进行聚类(例如,参见pmid:11294794);
步骤3:除去覆盖率相对于原始多肽链(目标蛋白)的覆盖率低于40%且序列同一性小于15%的序列;
步骤4:检查从步骤3产生的列表中每个序列的注释和来源生物,并排除很可能成为假阳性的序列。非限制性实例是没有分子-功能注释的命中物(通常将这些注释为“假设蛋白”),来自除了目标蛋白的属或门以外的属或门的序列,或注释为具有的功能不同于目标蛋白的功能的蛋白;
步骤5排除与原始多肽链(参见,例如,pmid:18048315)的成对比对中具有大于5%、大于4%、大于3%、大于2%、大于1%或大于0.5%的缺口(插入或缺失,通过缩写词indel已知)的序列;
步骤6:将从步骤5产生的序列与使用任何序列同一性搜索方案收集和处理的高序列同一性序列(即与目标蛋白的序列同一性超过30%)组合,并生成多序列比对(msa)。然后,即使该msa含有很少(少于3-10个)序列,其也可以通过本文呈现的方法用作输入。
以下是一个更具体但非限制性的实例:
步骤i:使用csi-blast搜索算法而不是blastp来鉴定同源物。在一些情况下,使用替代序列搜索算法来发现遥远的同源物(诸如使用具有3次迭代的csi-blast(背景特异性的迭代blast)替代blastp)是有利的,因为csi-blast构建了不同的取代矩阵来计算比对评分。csi-blast矩阵是背景特异性的(即,每个位置概率还取决于12个相邻氨基酸),因此在相同的错误率下,它发现的同源序列比blast多50%。迭代使用意味着重复该过程,并在每轮结束时根据来自直至该点收集到的同源物的序列信息更新取代矩阵。
步骤ii:分别使用19%和15%的最小序列同一性阈值用于严格比对和容许比对。在使用blastp的同时将最小序列同一性阈值降低至15%(容许比对)和19%(严格比对)可能是没有意义的,因为对blastp进行调谐以找到与靶标具有更高序列同一性的序列。其次,根据从csi-blast搜索获得的结果选择这些阈值;因此,这些阈值在csi-blast搜索后设置,并取决于结果;具体地,可能需要调整阈值以获得更多的真阳性或更少的假阳性结果,其中真阳性是具有功能注释和系统发生起源的命中物,其对应于下面的步骤iii的要求。
步骤iii:如果预期蛋白靶标的折叠或功能对于靶标蛋白的门的属是独特的,则排除来自除了对应于目标蛋白的属或门以外的属或门的序列。如果保持这种期望,则来自靶标蛋白的属和门之外的属和门的蛋白可能是假阳性命中物;即采用不同的折叠或功能的蛋白。
步骤iv:对于在与查询的成对比对中共有低于19%序列同一性的序列,使用高达1%的indel分数。在缺口/indel的处理中,对于具有低于19%的最小%同一性的序列,可能需要csi-blast成对比对indels分数高达1%。基本原理是,对于与查询共有这种小序列同一性的低同源性序列,在msa中插入假阳性的风险太高,但是小indel分数表明这些可能是真实命中物。
步骤v:使用命中物在比对中相对于靶标蛋白的序列覆盖率阈值为50%。通过步骤ii、iii和iv中列出的标准的所有序列可能表现出超过50%的覆盖率;然而,如本领域通常所实践,如果覆盖率阈值被设置为60%,则大多数序列将被滤掉。
步骤vi:如本领域通常所实践,为剩余序列生成msa。
可变的环区域:
blast算法可提供包括具有不同长度的序列的结果。差异通常源于环区域的不同长度,并且具有不同长度的环可反映不同的生物化学背景。作为结果,代表环位置的msa栏可包含来自具有不同长度的环的比对的残基,因此可能使具有来自不同生物化学背景的信息的数据降级(degrade),可能与目标蛋白的生物化学背景不相关。blast命中物因此在一些位置可包含相关信息,同时在其他位置包含不相关的信息。为了使对于每一环的不相关序列信息的水平最小化,鉴定原始蛋白的二级结构并且对于每一环区域创建背景特异性子msa文件,并且子msa仅包含具有相同长度的环序列。
二级结构鉴定通过鉴定结构中的氢键模式进行,并且这被称为“蛋白二级结构词典”(dssp)。存在几个提供这种分析的可用的软件包,诸如例如,用于环鉴定的rosetta™模块。
二级结构鉴定程序的输出通常为与模板结构具有相同长度的字符串(即,输出字符串),其中每一字符代表二级结构元件中的一个残基,其可为h、e或l,表示形成α-螺旋、β-折叠或环的一部分的氨基酸。
根据本发明的一些实施方案,原始蛋白的结构中环区域的氨基酸序列如下处理:
(a)模板结构中的环通过自动或手动检查结构模型,和/或通过任何二级结构分析算法鉴定。
(b)确定输出的字符串上代表每一环的位置,包括环茎(在环的每一末端处两个额外的氨基酸)。为了导致茎,向每一环的末端添加两个位置,除非环位于主链末端之一。根据本发明的一些实施方案,在环定义中包括茎是有利的,这是因为锚定不同环的茎可潜在地表现出不同的构象并在其自身之间或与环残基形成不同的接触,并且有利的是用作所呈现方法中的输入的序列数据将会代表它。
例如,如果二级结构输出字符串为:
则环区域定义在位置1-5、9-17和19-25(粗体字符)。
(c)在msa中在查询序列中鉴定代表每一环的位置。msa中的环位置可能与来自之前步骤的原始字符串中的环位置不同,因为在msa中,查询与其他序列比对,且因此可包含氨基酸字符和代表缺口的连字符二者。
(d)在msa中的查询序列中定位环位置后,对每一环定义字符模式。例如,模式可包含“x”字符以代表氨基酸以及“-”(连字符)代表缺口。
(e)最后,对于每一环生成背景特异性的子msa文件,排除对于该环不共享相同字符模式的所有序列,即背景特异性子msa包含其中环具有相同长度的序列,包括缺口。
例如,在假定的原始蛋白中位置4-10被识别为环,具有假定序列“aptesvv”,包括茎。将该环在msa文件中的查询蛋白上鉴定,并且发现模式为“a--ptesvv”。背景特异性子msa文件(其将用msa文件中的所有序列针对该环生成)将含有模式“x--xxxxx”。
因此,根据本发明的一些实施方案,对于环区域,序列比对包括具有等于原始多肽链中的相应环的序列长度的氨基酸序列。相应地,在环区域的背景中相关的序列比对在本文中称为“背景特异性子msa”。
取代规则:
该方法要求鉴定可取代残基。可取代残基的选择可能取决于专家指导的关于突变位置的决定。这些位置通常是酶的活性位点中对核心催化活性不是至关重要的、但在底物附近(第一壳)或在第一壳位置附近(第二壳)等的位置。
在本发明的一些实施方案中,一组抑制、约束和权重用作支配一些计算程序的规则。在本发明一些实施方案的背景下,这些规则应用于本文呈现的方法中以确定原始多肽链中的哪些位置将被允许重新排列(被取代),以及重新排列为哪个氨基酸替代物。这些规则也可用于保存,至少一些程度上,原始多肽链的序列中的一些位置。
氨基酸序列变更中利用的规则之一源于特定位置处高度保守的序列模式,其通常在结构上相似的蛋白的家族中表现出。根据本发明的一些实施方案,序列设计程序期间指定氨基酸取代的规则包括位置特异性评分矩阵值,或pssm。
“位置特异性评分矩阵”(pssm),在本领域中也称为位置权重矩阵(pwm),或位置特异性权重矩阵(pswm),是普遍使用的生物序列中重复出现模式的代表,其基于在沿着序列的给定位置处字符(单体;氨基酸;核酸等)出现的频率。因此,pssm代表在每一位置处观察突变为20种氨基酸中的任一种的对数似然。pssm经常来源自认为结构上和功能上相关的一组比对序列,并且已经广泛用在用于计算基序发现的许多软件工具中。在氨基酸序列的背景下,pssm为用于蛋白blast搜索中的一种类型的评分矩阵,其中对于蛋白多序列比对中的每一位置分别给出氨基酸取代评分。因此,比对的位置a处的tyr-trp取代可接收与位置b处同一取代非常不同的评分,受制于两个位置处不同的氨基酸保守水平。这与位置独立性矩阵、诸如pam和blosum矩阵不同,其中tyr-trp取代接收相同的评分,无论其在何位置发生。pssm评分一般显示为正或负的整数。正评分表明给定的氨基酸取代在比对中比偶然预计的更频繁地发生,而负评分表明该取代不如预计频繁地发生。大的正评分经常指示关键的功能残基,其可为活性位点残基或其他分子间或分子内相互作用所需的残基。pssm可使用位置特异性迭代基础局部比对搜索工具(position-specificiterativebasiclocalalignmentsearchtool)(psi-blast)[schäffer,a.a.等人,nucl.acidsres.,2001,29(14),pp.2994-3005]创建,其发现与查询序列相似的蛋白序列,且然后从所得的比对构建pssm。或者,pssm可从国家生物技术信息中心保守结构域数据库(thenationalcenterforbiotechnologyinformationconserveddomainsdatabase)(ncbicdd)数据库检索(retrieve),因为每一保守的结构域在种子比对中由编码观察到的取代的pssm代表。这些cd记录可通过在entrezconserveddomians中文本搜索或通过使用反向位置特异性blast(reverseposition-specificblast)(rps-blast)(也称为cd-搜索)找到,以在输入蛋白序列上定位这些结构域。
在本发明的一些实施方案的背景下,pssm数据文件可为整数的表格形式,各自表明在设计蛋白的序列中的任何可能的位置处20种氨基酸中的任一种如何进化上保守。如上文所表明,正整数表明氨基酸在给定的位置比其将会在随机蛋白中的随机位置更可能,并且负整数表明氨基酸在给定的位置不如其将会在随机蛋白中可能。一般而言,pssm评分根据输入msa中的信息和关于氨基酸取代本质上的一般信息的组合确定,如例如,通过blosum62矩阵所引入的[eddy,s.r.,natbiotechnol,2004,22(8),pp.1035-6]。
一般而言,本文呈现的方法可使用psi-blast软件包的pssm输出来获得原始msa和所有子msa文件两者的pssm。根据本发明的一些实施方案,最终的pssm输入文件包括来自每一pssm文件的相关行。对于代表二级结构的序列位置,相关行从源自原始全msa的pssm拷贝。对于每一环,相关行从源自代表该环的子msa文件的pssm拷贝。因此,根据本发明的一些实施方案,最终的pssm输入文件为序列数据的定量代表,其并入结构计算中,如下文所讨论。
根据本发明的一些实施方案,基于msa和pssm的规则确定原始多肽链的氨基酸序列中的不可取代的位置和可取代的位置,并且进一步确定哪些氨基酸替代物将充当所述方法的单一位置扫描步骤中的候选替代物,如下文所讨论。
关键残基:
根据本发明的一些实施方案,所述方法允许并入关于原始多肽链和/或野生型蛋白的信息。该信息可由各种来源提供,其作为设计程序期间支配氨基酸取代的规则的部分并入该方法中。虽然任选,但这种信息的加入是有利的,因为其减少所述方法提供包括折叠-和/或功能-消除取代的结果的可能性。在下面的实施例部分中呈现的实施例中,关于活性的有价值的信息已经被成功用作部分规则。
术语“关键残基”是指在规则中定义为至少某种程度上固定的(不可变的)的设计序列中的位置。关键残基占据的序列位置任选地构成不可取代的位置的一部分。
关于关键残基的信息可,例如,从原始多肽链的结构(或模板结构),或者当可用时从其他高度相似的结构提取。可帮助鉴定关键残基和支持推理在任何给定位置固定一种氨基酸类型或同一性的示例性标准,包括:
在先前提供的蛋白稳定性设计方法pross中,当用于提供稳定的酶变体时,关键残基在底物结合位点周围约5-8å的半径内选择,如可从包含底物、底物类似物、抑制剂等的复合晶体结构推断的。类似地,当使用pross来提供稳定的金属结合蛋白时,关键残基在金属原子周围约5-8å内选择。其他关键残基可在寡聚体中涉及目标链的蛋白界面中指定,因为相互作用链经常涉及二聚化界面、结合配体或蛋白-底物相互作用。同样,关键残基可以在距与目标蛋白相互作用的dna/rna链特定距离内、距表位区域特定距离内等指定。
应指出选择关键残基的空间的形状和大小不限于半径5-8å的球体;该空间可为对应于原始蛋白的序列、功能和结构的任何大小和形状。进一步指出的是具体的关键残基可由任何外部信息源(例如,研究者)提供。
在本发明的背景下,关键残基被少量选择(≤10个位置,且更通常为0-3个位置),甚至并且特别是在该方法尝试多样化或提高的活性区域内和周围。该策略允许活性确定区域多样化,同时不牺牲蛋白的稳定性。
当提供模板结构、pssm文件(其基于全msa和任何任选的背景特异性子msa)以及关键残基、不可取代的位置和可取代的位置的鉴定时,本文呈现的方法可使用这些数据从原始多肽链开始提供修饰的多肽链。
主要的方法步骤:
本文提供的方法(funclib/ablift)的目标是设计适用于低通量实验测试的稳定、有效且功能多样的多点活性位点突变体的小集合。设计策略是通用的,并且原则上可以使用其分子结构和同源序列的多样集合应用于任何天然酶或设计的蛋白。
根据本发明的一些实施方案,本文呈现的方法包括如下步骤,其确定原始多肽链的氨基酸序列中哪些位置将进行氨基酸取代以及将评价哪些氨基酸替代物(在本文中称为可取代位置),并且其中原始多肽链的氨基酸序列中的哪些氨基酸位置将不进行氨基酸取代(在本文中称为不可取代位置)。
在下一步骤,(单一位置扫描步骤),向每一可取代的位置处的每一允许的氨基酸替代物给予位置特异性稳定性评分。在酶库情况下,活性位点残基被定义为通过视觉检查酶分子结构来设计。从pssm计算进化保守性评分,并且基本上如先前所述[goldenzweig,a.等人molcell.,2016,63(2),pp.337-346]计算δδg值。根据以下阈值过滤在pte的活性位点处的耐受的氨基酸身份:pssm≥-2和δδg≤+6r.e.u。
应注意,本文呈现的方法的详细描述使用rosetta™软件包所共有或独特的一些项、单元和程序,然而,应理解所述方法能够使用其他软件模块和包执行,并且其他项、单元和程序因此考虑在本发明的范围内。
还应注意,本文呈现的方法的详细描述使用实施例部分中呈现的蛋白和变量,其不应被视为以任何方式进行限制,因为该方法适用于所需数据可用的任何蛋白和多肽链序列。
根据本发明的一些实施方案,该方法的以下步骤是穷举列举原始多肽链(例如pte)中的至少3个以及多达5、6、7、8、9、10或更多个六个突变的所有可能的组合。每种突变体都在rosetta中建模,包括组合侧链堆积,并且使所有残基的骨架和侧链能量最小化,在整个蛋白(由一条或多条多肽链构成)的cα坐标上受到谐波抑制。根据所有原子能量对所有设计的多肽链(设计的蛋白或简称为“设计”)进行排序,并且在除去相对于彼此具有少于两个突变的设计后,选择排序靠前的设计用于实验分析。
如上文所述,pross和本文呈现的方法之间的主要差异之一是pross中的组合设计步骤,其被本方法中的综合列举步骤替代。在此处呈现的示例性研究中,对本文呈现的方法(funclib/ablift)进行的小规模测试证明足以鉴定表现出酶活性概况的数量级变化而不损失表观蛋白稳定性的变体。因此,所述方法可用于快速优化特定活性或从不适合高通量筛选的酶生成功能库。常规的活性位点设计策略依赖于过渡态建模,而本文提供的方法计算相互作用的活性位点突变的多样且稳定的网络,使得甚至在此处讨论的情况下(其中酶过渡状态模型不确定)进行设计。尽管设计的突变保留野生型骨架结构,但一些设计表现出标志上位性关系,这使得这些设计几乎无法进入逐步突变轨迹。因此,酶活性位点的序列空间提供了丰富的功能多样性资源,其无法通过自然和实验室进化来探索,但现在可以通过计算蛋白设计来访问。
根据本发明的一些实施方案,所述方法对于包含超过100个氨基酸(aa)的原始多肽链有效执行。在一些实施方案中,原始多肽链包含超过110aa、超过120aa、超过130aa、超过140aa、超过150aa、超过160aa、超过170aa、超过180aa、超过190aa、超过200aa、超过210aa、超过220aa、超过230aa、超过240aa、超过250aa、超过260aa、超过270aa、超过280aa、超过290aa、超过300aa、超过350aa、超过400aa、超过450aa、超过500aa、超过550aa或超过600个氨基酸。
根据本发明的一些实施方案,本文呈现的方法提供与开始的原始多肽链相比具有超过2个氨基酸取代(突变)、超过3个取代、超过4个取代、超过5个氨基酸取代、超过6个取代、超过7个取代、超过8个取代、超过9个取代、超过10个取代、超过11个取代或超过12个取代的修饰的多肽链。
序列空间:
根据本发明的一些实施方案,过滤关键残基和强加自由能接受阈值之后,给定序列中可取代位置的数目大大减少,由此提供宽但可管理的组合序列空间,可从其中选择设计序列。因此,术语“序列空间”是指一组可取代的位置,其各自具有相对于给定位置处的原始/wt氨基酸的至少一个任选取代。
序列空间因此为某一接受阈值的结果;每一接受阈值产生不同的序列空间,其中由较严格的接受阈值定义的序列空间包含在由更容许的接受阈值定义的更大的序列空间内。如上文所讨论,为了避免假阳性,接受阈值可为小的并且应为负的,其中-2r.e.u被认为是高度限制的(严格的),且+6r.e.u为高度容许的。通过使用接受阈值+6r.e.u获得的序列空间将必然比通过使用接受阈值-2.00r.e.u(严格的)获得的序列空间更大(容许)。实验使用本文呈现的方法以产生实际蛋白已经显示中间的接受阈值产生最佳的序列空间。实际上,序列空间为由pssm规则定义的更宽空间的亚空间。
呈现序列空间的示例性和一般的方法为基于野生型序列编号p1,p2,p3,…,pn的序列位置的列表,其中每一位置被指定为关键残基,即wt中发现的氨基酸,aawt;或可基于pssm和能量最小化分析采用来自包含至少一个替代氨基酸的有限列表的任何一个氨基酸的位置,aam,其中m为表示一个天然存在的氨基酸的数字,例如,a=1,r=2,n=3,d=4,c=5,q=6,e=7,g=8,h=9,l=10,i=11,k=12,m=13,f=14,p=15,s=16,t=17,w=18,y=19和v=20(aa编号为任意的,并且在本文中用于表明序列空间的一般代表)。
例如,序列空间可呈现为:
p1:aawt、aa5、aa8和aa12;
p2:aawt;
p3:aawt和aa16;
p4:aawt、aa1、aa3、aa6、aa10和aa14;
p5:aawt、aa4、aa8和aa11;
…
pn:aawt、aam、aam、aam、aam和aam,;
其中在该一般性的实例中,p1具有4个替代氨基酸,p2为关键残基,等。
根据本发明的一些实施方案,序列空间可进一步通过强加更严格的接受阈值限制,或通过强加更容许的接受阈值扩展。一般而言,发现值+2r.e.u为足够容许的;然而也考虑基于大于+2r.e.u的接受阈值(例如,+6r.e.u)或基于小于-2.00r.e.u的接受阈值(例如,-2.1r.e.u)的序列空间。
在下面随后的实施例部分中,对于已经证明所述方法的一些示例性蛋白,呈现了基于接受阈值+6r.e.u的序列空间。具有选自呈现的序列空间的相对于野生型/起始序列的任何2个或更多个取代的选择并且表现出至少一种提高的催化活性的任何设计序列考虑在本发明的范围内。
本文注意到本发明的实施方案包括由本文呈现的方法提供的任何给定的序列空间中的氨基酸替代物的任何和所有可能组合(源自如本文所定义的序列空间的所有可能的变体)。
进一步注意到,在本发明的一些实施方案中,由在原始蛋白上执行本文呈现的方法而产生的序列空间可应用于与原始蛋白不同的另一个蛋白,只要其他蛋白表现出至少30%、至少40%或至少50%序列同一性以及更高。例如,取自通过在人蛋白上执行本文呈现的方法所提供的序列空间的一组氨基酸替代物可用于通过产生在序列等效位置具有氨基酸取代的非人蛋白的变体来修饰非人蛋白。所得的非人蛋白的变体,本文中称为“杂合变体”,然后将会在与人蛋白中的相应位置对齐的位置具有“人氨基酸取代”(选自为人蛋白而提供的序列空间)。在本发明的一些实施方案中,具有匹配本文呈现的方法所提供的任何给定序列空间中的氨基酸替代物的至少2个取代的任何这样的杂合变体(源自如本文所定义的序列空间的所有可能的变体),考虑并包括在本发明的范围内。
funclib网络服务器:
构建funclibweb服务器以实现本文呈现的方法的几项改进。在设计示例性酶pte变体中,如本文所呈现,针对整个蛋白序列计算多序列比对(msa),并且在查询结构中观察到任何环的地方,消除表现出相对于查询的缺口的任何比对序列以减少比对不明确(参见[goldenzweig,a.等人.molcell.,2016,63(2),pp.337-346])。相比之下,在funclib网络服务器中,所有二级结构元件都进行该过滤,导致pssm准确性提高,尤其是在活性位点口袋中。此外,网络服务器实现更准确的原子建模和评分:其使用近来的rosetta能量函数[park,h.等人,jchemtheorycomput.,2016,12(12),pp.6201-6212],相对于先前的rosetta能量函数,其静电和溶剂化潜能提高;对催化口袋中的必需氨基酸残基的侧链原子实施谐波坐标抑制,以确保其预先组织;将精修限制在设计位置的8å内(或6-10å的范围内)的氨基酸,而不是精修整个蛋白;允许用户修改耐受的序列空间(例如,基于先前的实验和结构分析);并使得能够对小分子配体或过渡态复合物进行建模。
多样磷酸三酯酶库:
改变活性的自然和实验室进化取决于突变的逐步积累,每个突变在适应性上必须至少是中性的。然而,在几个突变后,由于上位性或稳定性-阈值效应,活性的提高经常趋于平稳。因此,导致从一种高效酶到另一种高效酶的典型进化轨迹是耗时的,并且经常在活性位点之外包含数十种实现突变,其中大多数仅间接地例如通过稳定酶来对活性有贡献。本文呈现的策略合理化并加速表现出改变活性的稳定酶的生成:其通过使用先前提供的方法(pross)设计稳定且高度表达的酶变体而开始,且然后设计数十种变体,所述变体编码表现出不同的立体化学特征的活性位点突变体的预先组织的网络。进化保守性分析和rosetta原子建模的组合使设计计算聚焦于稳定、预先组织和功能性的活性位点集合。
因此,本发明人已经实施了funclib程序,以便列举对底物具有增强的催化活性的pte变体,而wtpte对所述底物的催化活性不太有效,因为此类pte变体可以充当针对各种有机磷酸酯/神经试剂的解毒剂,以及增加pte对已知pte底物(诸如vx型神经试剂)的催化活性。使用pross-稳定的序列[wo2017/017673;goldenzweig,a.等人.molcell.,2016,63(2),pp.337-346]dpte2(seqidno:1)(其为pte的变体,其在活性位点口袋之外含有20个突变且源自pte-s5[roodveldt,c.和tawfik,d.s.,proteinengdessel.,2005,18(1),pp.51-8]),并使用wtpte(pdb条目:1hzy)的晶体结构,通过本文呈现的方法获得的设计变体表现出相对于wtpte具有数千倍活性的广谱活性。
因此,根据本发明的一个方面,提供了具有选自对于作为原始蛋白的来自缺陷假单胞菌的磷酸三酯酶(pte)提供且在表a中列出的序列空间的至少2个氨基酸取代的任何组合的序列的蛋白,而其中未显示野生型位置i106、f132、h254、h257、l271、l303、f306和m317。
根据本发明的一些实施方案,所述蛋白可以选自本文列出的表a中呈现的列表。在一些实施方案中,所述蛋白具有选自以下的序列:pte_28(seqidno:28)、pte_29(seqidno:29)、pte_56(seqidno:56)和pte_57(seqidno:57)。
根据一些实施方案,所述蛋白可以是分离的蛋白,与另一结构域(诸如fc)的融合体,或蛋白和其他试剂、因子载体等的混合物,只要其包括如表a中定义的pte设计的蛋白中的至少一种。
所述原始蛋白可以是具有ecno.3.1.8.1(ec:3.1.8.1)的pte家族的任何酶,包括来自缺陷假单胞菌或任何其他生物的野生型pte,或任何设计的人工pte,包括通过使用诸如但不限于pross的计算方法获得的pte变体。为了鉴定取代任何原始蛋白的氨基酸残基,将原始蛋白的序列与如pdb条目:1hzy中所呈现的来自缺陷假单胞菌的磷酸三酯酶(pte)的序列进行比对。
如本文所用,本文缩写为pte的术语“磷酸三酯酶”(也称为对硫磷水解酶(ec:3.1.8.1))是指属于酰胺水解酶超家族的酶。本发明的该方面的磷酸三酯酶是细菌磷酸三酯酶,与其他磷酸三酯酶相比,由于延长的环7氨基酸序列,其对v-型有机膦酸酯具有增强的催化活性。此类磷酸三酯酶已在缺陷短波单胞菌、黄杆菌属(pteflavob)和农杆菌属中鉴定。
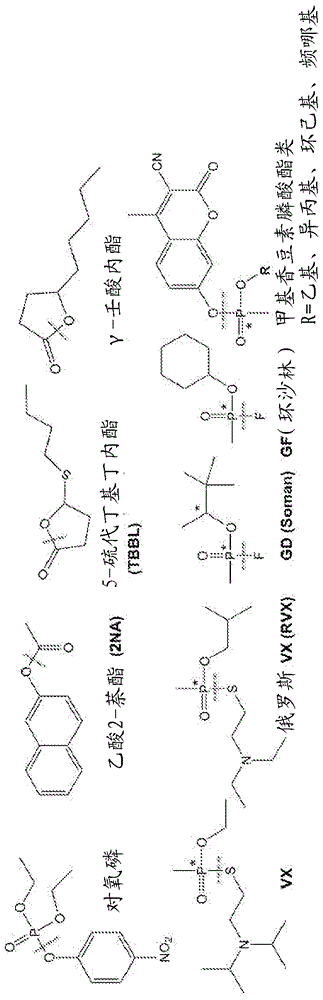
如本文所用,“神经试剂”是指诸如具有乙酰胆碱酯酶抑制活性的有机磷酸酯(op)化合物。op化合物的毒性取决于其对乙酰胆碱酯酶的抑制速率以及离去基团(诸如氟化物、烷基硫醇化物、氰化物或芳氧基)的伴随释放。神经试剂可以是外消旋组合物或纯化的对映异构体(例如,sp或rp)。在本发明的实施方案的背景下,术语“有机磷酸酯”或“神经试剂”涵盖v-型(amiton)神经试剂、g-型(trilon)神经试剂和gv-型(novichok)神经试剂。在本发明的实施方案的背景下,术语“神经试剂”包括但不限于g-型试剂,诸如tabun(ga)、sarin(gb)、chlorosarin(gc)、soman(gd)、ethylsarin(ge)和环沙林(gf),v-型试剂,诸如ea-3148、ve、vg、vm、vp、vr、vs、r/s-vx、cvx和rvx,以及gv-型试剂,诸如novichok试剂和gv(2-[二甲基氨基(氟)磷酰基]-n,n-二甲基乙胺)。
有机磷酸酯解毒的方法:
根据本发明的一个方面,本文提供的设计的蛋白或pte变体可用于通过如下将设备、衣服和环境去污:水解广谱的有机磷酸酯试剂(包括来自g-型、v-型和gv型神经试剂的神经试剂),且由此使怀疑被此类试剂污染的物体或区域解毒。该区域可以是无生命的物体、地面、一片设备、一片衣服和身体表面。
在一些实施方案中,本文提供的设计的蛋白或pte变体可以在体内施用于怀疑神经试剂中毒的受试者。在此类用途中,所述蛋白作为药物组合物施用,并且可以包括药学上可接受的载体以及其他活性成分和赋形剂。
预期在本申请到期的专利有效期期间,将开发许多具有有机磷酸酯的广泛特异性水解的相关设计的pte变体,并且短语“设计的pte变体”的范围意欲先验地包含所有此类新技术。
如本文所用,术语“约”是指±10%。
术语“包含(comprises)”、“包含(comprising)”、“包括(includes)”、“包括(including)”、“具有”及其缀合物意指“包括但不限于”。
术语“由……组成”意指“包括且限于”。
如本文所用,在某种物质的背景下,短语“实质上没有”和/或“基本上没有”是指完全没有该物质或以组合物的总重量或体积计包括小于约5、1、0.5或0.1%的该物质的组合物。或者,在过程、方法、特性或特征的背景下,短语“实质上没有”和/或“基本上没有”是指完全没有特定过程/方法步骤的过程、组合物、结构或制品,或者特定特性或特定特征或其中特定过程/方法步骤与给定标准过程/方法相比小于约5%、1%、0.5%或0.1%受影响的过程/方法,或者特征在于与给定标准相比小于约5%、1%、0.5%或0.1%的特性或特征受影响的特性或特征。
如本文所用,单数形式“一个/种(a)”、“一个/种(an)”和“该/所述”包括复数指示物,除非上下文另有明确说明。例如,术语“一种化合物”或“至少一种化合物”可以包括多种化合物,包括其混合物。
本申请自始至终,本发明的各个实施方案可以以范围形式呈现。应当理解以范围形式的描述仅为了方便和简洁起见,并且不应解释为对本发明的范围的硬性限制。相应地,范围的描述应视为已具体公开在该范围内的所有可能的子范围以及个别数值。例如,范围的描述诸如1至6应视为已具体公开在该范围内的子范围诸如1至3、1至4、1至5、2至4、2至6、3至6等,以及在该范围内的个别数目,例如1、2、3、4、5和6。这与范围的宽度无关应用。
每当本文指出数目范围时,它意欲包括在所述范围内的任何引用数目(分数或整数)。短语“范围在第一指示数目和第二指示数目……之间/在第一指示数目和第二指示数目……之间的范围”和“范围从第一指示数目到第二指示数目/从第一指示数目到第二指示数目的范围”在本文中可互换使用,并且意欲包括第一和第二指示数目,以及两者之间的所有分数和整数数目。
如本文所用,术语“方法”是指用于完成给定任务的方式、手段、技术和程序,包括,但不限于,化学、药理学、生物学、生物化学和医疗领域的从业者所已知的或从已知的方式、手段、技术和程序容易地开发的那些方式、手段、技术和程序。
如本文所用,术语“治疗”包括消除、实质上抑制、减缓或逆转病况进展、实质上改善病况的临床或美学症状或者实质上防止病况的临床或美学症状出现。
当参考具体的序列表时,这种参考应理解为也包括实质上对应于其互补序列的序列,包括较少的序列变异,由例如,测序错误、克隆错误或者导致碱基取代、碱基缺失或碱基添加的其他变异导致,条件是此类变异的频率小于50个核苷酸中1个,或者,小于100个核苷酸中1个,或者,小于200个核苷酸中1个,或者,小于500个核苷酸中1个,或者,小于1,000个核苷酸中1个,或者,小于5,000个核苷酸中1个,或者,小于10,000个核苷酸中1个。
应当理解,本申请中公开的任何序列标识号(seqidno)可以指dna序列或rna序列,这取决于其中提及该seqidno的背景,即使仅以dna序列格式或rna序列格式表示该seqidno。例如,seqidno:#以dna序列格式表示(例如,以t表示胸腺嘧啶),但它可以指对应于#核酸序列的dna序列,或rna分子核酸序列的rna序列。类似地,尽管一些序列以rna序列格式表示(例如,以u表示尿嘧啶),这取决于所描述分子的实际类型,但它可以指包含dsrna的rna分子的序列,或对应于所示rna序列的dna分子的序列。在任何情况下,设想具有公开有任何取代基的序列的dna和rna分子两者。
应当理解,为了明确起见,在单独的实施方案的背景下描述的本发明的某些特征也可以在单个实施方案中组合提供。相反,为了简洁起见,在单个实施方案的背景下描述的本发明的各种特征也可以单独或以任何合适的子组合或如本发明的任何其他所述实施方案中合适的提供。在各个实施方案的背景下描述的某些特征不视为那些实施方案的基本特征,除非实施方案没有那些要素则不起作用。
如在上文描绘和如下文权利要求部分中请求保护的本发明的各个实施方案和方面可在下述实施例中找到实验或计算支持。
实施例
现在参考下述实施例,所述实施例连同上文说明书一起以非限制性方式举例说明本发明的一些实施方案。
实施例1
计算方法
本平台的实施方案,也称为funclib,旨在设计适用于低通量实验测试的一小组稳定、有效且功能多样的多点活性位点突变体。设计策略是通用的,并且原则上可以使用其分子结构和同源序列的多样组应用于任何天然酶(图1a-d)。
计算工具:
用于生物分子设计的rosetta软件套件用作该方法的计算部分的框架,并且可用于在www(dot)rosettacommons(dot)org下载。具体地,rosettagithub版本627f7dd22223c3074594934b789abb4f4e2e3b10用于所有模拟。所有rosetta建模和设计都使用rosettascripts[fleishman,s.l.等人,plosone,2011,6(6)](其用下文的其命令行和标记文件得到)进行。所有设计计算都使用rosettatalaris14全原子能量函数,其主要由范德华堆积、氢键键合、溶剂化和静电作用主导。
funclib设计策略:
本文提供的方法(funclib)的目标是设计适用于低通量实验测试的一小组稳定、有效且功能多样的多点活性位点变体(突变体)。使用的设计策略是通用的,并且可以使用其分子结构和同源序列的多样组应用于任何天然酶或设计的蛋白。
图1a-c呈现示意性流程图,其举例说明用于产生给定酶的功能设计的文库的方法中的关键步骤。仅例如且非限制性地,图1a-c举例说明从细菌磷酸三酯酶(pte;pdb条目:1hzy)的晶体结构以及pte,dpte2(seqidno:1)的pross-稳定的变体的序列开始生成磷酸三酯酶(pte)酶库中的步骤。具体地,图1a显示这样的步骤,其中选择活性位点位置用于设计,并且在每个位置,序列空间受到进化保守分析(pssm)和突变扫描计算(δδg)的约束。图1b显示其中使用rosetta原子设计计算详尽列举多点突变体的步骤。在出于证实目的呈现的实例中,pte活性位点包含zn2+离子的双金属中心(灰色球体),其被六个高度保守的残基(灰色棒)配位;八个额外的残基(有色棒)包含活性中心壁并且保守性较低。图1c显示其中根据能量对设计进行排序的步骤,且图1d显示其中将序列聚类以获得用于实验测试的多样化、低能量设计的库的步骤。设计位置在图1a-c始终一致地着色。
如图1c中所见,基于上文呈现的规则,对每一设计的结构进行全局能量最小化,并确定每一设计结构相对于模板结构的总自由能的最小化能量评分。根据本发明的一些实施方案,将设计结构根据最小化能量评分进行分选。
为了证明本文呈现的方法从缺陷假单胞菌选择金属酶磷酸三酯酶(pte)的原因之一是,除了高效水解有机磷酸酯杀虫剂对氧磷(kcat/km近似108m-1s-1)以外,pte混杂水解酯、内酯和各种有机磷酸酯,包括毒性神经试剂,诸如vx、俄罗斯vx、soman(gd)和环沙林(gf),尽管kcat/km值比对氧磷低几个数量级。
然而,用于体内保护的有效的有机磷酸酯解毒需要高催化效率,其中最小kcat/km为107m-1min-1,由此激发靶向pte的几种近来的酶工程改造尝试。此外,来自结构类似于vx和gf的新一代神经试剂(“novichoks”)的威胁加强了对广谱神经试剂水解酶的需求。
图2a-c呈现使用根据本发明的实施方案的funclib方法的一些结果,其中设计的磷酸三酯酶(pte)库表现出一定范围混杂活性的数量级的提高。具体地,图2a显示细菌pte是对氧磷酶,其表现出额外的混杂水解酶活性,其中虚线指示在该研究中测试的每种底物中pte水解的键,且星号指示手性中心。图2b显示顶级funclib设计的催化效率(kcat/km)相对于pte-s5的x倍提高,显示在几种设计中神经试剂水解效率显著提高>1,000倍,而活性位点突变的数目在条上方指示。图2c显示顶部pte设计的活性概况,其中几种设计,最主要pte_28(seqidno:28)、pte_29(seqidno:29)和pte_56(seqidno:56),表现出相对于原始序列的酶显著拓宽的底物选择性。对于毒性更强的sp立体异构体显示神经试剂的数据。数据表示为一式两份测量值的平均值±标准偏差;n.d.-未测定。图2b的x-轴中的数字和图2c中的y-轴中的数字代表变体编号(pte_x)和seqidno:x)。
由于活性位点突变经常损害蛋白稳定性,因此活性位点设计计算可以从原始多肽链的稳定设计(即通过诸如pross的方法提供的设计(参见上文))的多肽链开始。在用于证明本文提供的方法的实例中,发明人采用dpte2(seqidno:1),其为pte-s5的变体[roodveldt,c.和tawfik,d.s.,proteinengdessel.,2005,18(1),pp.51-8],其中活性位点口袋之外的20个稳定突变先前使用pross稳定性-设计算法设计[goldenzweig,a.等人.molcell.,2016,63(2),pp.337-346]。原始序列dpte2(seqidno:1)表现出比pte-s5更高的稳定性和高五倍的细菌表达产率,同时保留野生型活性水平。
选择构成pte活性位点壁(第一壳)的八个活性位点位置用于设计方法,然而,应注意,起始位置的数量取决于方法的主题及其可用信息而不同。使用funclib的方法通过定义序列空间而开始,所述序列空间包含被预测为可以单独耐受的活性位点点突变(参见图1a)。首先,仅保留根据同源物的多序列比对在自然多样性中具有至少中等发生概率的突变。其次,消除根据rosetta原子建模实质上使原始序列(在本文中和通篇也称为“野生型”;“起始模型”;“原始结构”;或“模板序列”)不稳定的点突变。应用于pte活性位点口袋,在其zn2+螯合残基(不可取代或固定位置)中不允许突变,而在其他第一壳位置(可取代位置)允许,甚至是自由基突变(参见图1a-b)。如果在每个位置允许所有20种氨基酸,则两步过滤将多点突变体在8个活性位点位置处的组合空间从1010个突变体急剧减少至<105。从该过滤组,相对于原始序列包含3-5个突变的所有多点突变体均在rosetta中建模和精修,包括骨架和侧链最小化(参见图1b)。其后,根据其预测的稳定性对所有多点突变体进行排序(参见图1c)。因此,预测排序靠前的设计表现出稳定且预先组织的活性位点口袋-高催化效率的前提条件。令人惊讶地,发现数百种独特的活性位点设计表现出的能量评分比pte的起始序列的能量评分有利或更好,表明可通过计算设计接近活性位点处的潜在耐受的多点突变体的非常大的空间。根据一些实施方案,所述方法进一步包括这样的步骤,其中将设计聚类(参见图1d),由此消除与彼此或与野生型相差少于两个活性位点突变的设计。在这项使用pte的示例性研究中,选择49种靠前的设计用于体外实验(参见表1)。
方法结果和序列空间:
表1呈现使用如上文所述的funclib从pte的原始序列dpte2(seqidno:1)开始获得的结果,并且至少在某种程度上代表设计用于改进对广谱底物的反应性的pte变体的序列空间。粗体标记的是变体pte_28(seqidno:28)、pte_29(seqidno:29)、pte_56(seqidno:56)和pte_57(seqidno:57),其表现出相对于原始序列的酶显著拓宽的底物选择性。
rosettascriptsxml和标记文件:
精修
实施例2
功能文库制备
材料:
底物如先前所公开合成:5-硫代丁基丁内酯(tbbl)[khersonsky,o.和tawfik,d.s.,chembiochem,2006,7,pp.49–53];具有氰基香豆素离去基团的膦酸酯,乙基甲基磷酸氰基香豆素(emp),异丙基甲基磷酸氰基香豆素(imp),环己基甲基磷酸氰基香豆素(cmp)和频哪基甲基磷酸氰基香豆素(pmp)[ashani,y.等人,chemico-biologicalinteractions,2010,187(1-3),pp.362–369];和vx和rvx对映异构体[berman,h.a.和leonard,k.,j.biol.chem.,1989,264,pp.3942–3950]。
所有其他试剂(对氧磷、马拉息昂、乙酸对硝基苯酯、辛酸对硝基苯酯、乙酸2-萘酯、γ-壬酸内酯、dtnb、间甲酚、乙酸钠、丙酸、丁酸、异丁酸、戊酸、异戊酸、乳酸钠、己酸、nadh、乳酸脱氢酶、磷酸烯醇丙酮酸、丙酮酸激酶、3-磷酸腺苷、辅酶a)购自sigma-aldrich,且酵母肌激酶购自merck。
克隆:
将原始酶和设计变体的合成基因进行密码子优化,用于有效大肠杆菌表达,并由twistbioscience定制合成为线性片段。扩增pte设计的基因,并通过ecori和psti限制性位点克隆至具有n-末端mbp融合标签的pmalc2载体中。将质粒转化至大肠杆菌bl21de3细胞中,并提取dna用于sanger测序以验证准确性。具有活性设计的基因的质粒保存在addgene(保存号75507)。
蛋白表达:
将2ml的补充有100μg/ml氨苄青霉素(和在pte情况下的0.1mmzncl2)的2yt培养基用单个菌落接种,并在37℃下生长约15小时。将10ml补充有50μg/ml卡那霉素(和在pte情况下的0.1mmzncl2)的2yt培养基用0.2ml过夜培养物接种,并在37℃下生长至约0.6的od600。用0.2mmiptg诱导过表达,并使培养物在20℃下生长约24小时。离心并在-20℃下储存后,将沉淀重悬浮于裂解缓冲液中,并通过超声处理进行裂解。
pte纯化:
pte裂解缓冲液:50mmtris(ph8.0),100mmnacl,10mmnahco3,0.1mmzncl2,benzonase和0.1mg/ml溶菌酶。蛋白与直链淀粉树脂(neb)结合,用含有100mmnacl和0.1mmzncl2的50mmtris洗涤,并用含有10mm麦芽糖的洗涤缓冲液洗脱蛋白。洗脱级分用于sds-page凝胶,并且在活性测定之前,将蛋白在洗涤缓冲液中透析。为了结晶,将pte变体重新克隆至含有n-末端6xhis标签和mbp融合体的petmbph载体中[peleg,y.和unger,t.,methodsmol.biol.,2008,426,pp.197–208],并用500ml培养物进行表达。在纯化后,将蛋白用tev蛋白酶消化,以除去mbp融合标签(1:20tev,1mmdtt,24-48h/rt)。通过与ni2+-nta树脂结合除去mbp融合体,并通过凝胶过滤(hiload26/600superdex75制备级柱,ge)纯化蛋白。
动力学测量:
在活性缓冲液(含有100mmnacl和0.1mmzncl2的50mmtrisph8.0)中用纯化的蛋白进行pte设计的动力学测量。根据活性,使用一定范围的酶浓度。pte设计的活性用磷酸三酯类(对氧磷(0.5mm)、马拉息昂(0.25mm)、emp、imp、cmp、pmp(各0.1mm)、酯类(乙酸对硝基苯酯(0.5mm)、辛酸对硝基苯酯(0.1mm)、乙酸2-萘酯(0.3mm)和内酯(tbbl)(0.5mm)、γ-壬酸内酯(0.5mm,ph-敏感性测定,通过监测577nm处间甲酚指示剂的吸光度)进行比色测试。在96-孔板(光学长度–0.5cm)中进行动力学测量,并减去背景水解速率。
如所述[cherny,i.等人,acschembiol.,2013,8(11),pp.2394-403]进行在有机磷酸酯(op)水解酶存在的情况下v-型神经试剂的水解速率。如先前所述[ashani,y.等人,toxicologyletters,2011,206,pp.24–28;和gupta,r.d.等人,natchembiol.,2011,7(2),pp.120-5],进行在稀释水溶液中香豆素替代物至相应的g神经试剂的原位转化以及通过op水解酶对g试剂的解毒速率的监测。注意,原位生成的g-和v-试剂的浓度最不危险,因为原位合成在稀释水溶液中以小(mg)规模进行。尽管如此,由于它们作为ache的抑制剂的高功效,因此严格遵守所有安全要求。
通过在michaelis-menten方程的近似一阶动力学区域中测量几种低底物浓度下的活性,测定最高活性的pte设计的催化效率(kcat/km)。所有报告的值都代表基于至少两次独立测量的平均值±标准偏差。
pte设计结构的结构确定和精修:
使用悬滴蒸气扩散方法用mosquito机器人(ttplabtech)获得pte_6(seqidno:6)、pte_28(seqidno:28)和pte_29(seqidno:29)的晶体。所有数据集都在内部rigakuru-h3rx-射线上在单晶上以100k收集。从0.85m硫酸锂和0.05mhepesph=7.0生长pte_6(seqidno:6)的晶体。在空间群p43212中形成晶体,每个不对称单元具有一个二聚体,并衍射至1.63å分辨率。从0.1mmgcl2*6h2o、10%peg4000和0.05mtrisph=7.5生长pte_28(seqidno:28)的晶体。在空间群c2中形成的晶体,每个不对称单元具有一个二聚体,并衍射至1.9å分辨率。从0.1mmg(oac)2*4h2o、8%peg8000和0.05m甲次砷酸钠ph=6.4生长pte_29(seqidno:29)的晶体。在空间群c2中形成晶体,每个不对称单元具有一个二聚体,并衍射至1.95å分辨率。
使用mosflm程序对pte_6(seqidno:6)、pte_28(seqidno:28)和pte_29(seqidno:29)晶体的衍射图像进行索引和积分,并使用scala程序对积分反射进行缩放。使用来自ccp4程序套件的truncate计算结构因子振幅。通过用程序phaser进行分子置换来解析pte_6(seqidno:6)、pte_28(seqidno:28)和pte_29(seqidno:29)结构。用于解析pte_6(seqidno:6)、pte_28(seqidno:28)和pte_29(seqidno:29)结构的模型是工程改造的有机磷水解酶(pdb条目:1qw7)。
原子精修的所有步骤都用ccp4/refmac5程序和通过phenix精修实施。通过使用coot程序将模型构建至2mfobs-dfcalc和mfobs-dfcalc图谱中。pte_6(seqidno:6)、pte_28(seqidno:28)和pte_29(seqidno:29)结构的精修统计的细节描述于表1中。pte_6(seqidno:6)、pte_28(seqidno:28)和pte_29(seqidno:29)的坐标分别保存在rcsb蛋白数据库中,登录号分别为6gbj、6gbk和6gbl。所述结构将在公开后发布。
实施例3
功能文库表征
所有pte设计都保留可检测到的对氧磷酶活性水平(参见下面的表2),表明尽管存在高序列多样性,但它们的活性位点是完整且有功能的。
pte变体和对氧磷/马拉息昂:
表2呈现pte变体对磷酸三酯类对氧磷(0.5mm)和马拉息昂(0.25mm)的比活性(对于mg蛋白,μm产物/min)。
用替代的混杂的底物来测量所述变体的比活性,所述底物包括除了对氧磷以外的磷酸三酯类、膦酸二酯类、羧基-酯类和内酯类(参见图2a)。在该初始筛选后,确定活性最高的设计的催化效率。大多数设计就至少一种底物而言表现出效率增加:10种设计在水解杀虫剂马拉息昂中表现出高达14倍的效率提高,15种设计显示内酯酶效率的类似水平的提高(高达16倍),且35种设计表现出酯酶效率的高达1,000倍的显著增加(参见图2b-c,表3和表5)。
pte变体和具有香豆素的磷酸三酯类:
表3呈现pte变体对于具有香豆素离去基团的磷酸三酯类(0.1mm)的比活性(对于mg蛋白,μm产物/min)。粗体指示松散的对映选择性(未观察到两种立体异构体的不同水解速率的双相行为特征)。
pte变体和酯类:
表4呈现pte变体对于酯类的比活性(对于mg蛋白,μm产物/min)。nd=低于检测限值。
pte变体和内酯类:
表5呈现pte变体对于内酯类的比活性(对于mg蛋白,μm产物/min)。nd=低于检测限值。
除了表现出提高的针对一定范围底物的催化效率以外,根据本发明的一些实施方案,本文呈现的pte变体还显示底物选择性的巨大变化。例如,pte-s5对对氧磷的选择性是酯乙酸2-萘酯(2na)的3x104-倍。仅通过五个活性位点突变,变体pte_37(seqidno:37)中的选择性被逆转为0.04;接近百万倍的选择性转变。类似地,pte-s5对对氧磷的有利性是合成内酯四丁基丁内酯(tbbl)的103倍,而在设计pte_27(seqidno:27)中,选择性被转变为0.1(参见,下表6)。
pte变体的催化效率:
表6呈现pte变体的特异性变化(作为催化效率的比率,kcat/km)。
值得注意的是,这些设计保留显著的对氧磷酶活性(kcat/km≥104m-1s-1),表明一些设计拓宽了底物识别,而不是仅针对另一种活性权衡一种活性(参见,图2c)。与该结论一致,相对于野生型,几种设计表现出就甲基香豆素膦酸酯的不利立体异构体而言增加的效率,同时保留针对天然有利的立体异构体的高效率(参见,表3)。
接下来,测量对于毒性神经试剂vx、俄罗斯vx(rvx)、soman(gd)和环沙林(gf)保留高磷酸三酯酶活性的设计的催化效率(参见,表7和表8)。
表7呈现pte变体对于v型的神经试剂的活性,kcat/kms-1m-1。
表8呈现最佳的pte设计对于神经试剂的活性与通过定向进化获得的pte变体对于神经试剂的活性的比较;kcat/km,x106m-1min-1,在含有50mmnacl的50mmtris中在ph8、25℃下测量。
a取自cherny等人[cherny,i.等人,acschembiol.,2013,8(11),pp.2394-403]的wt-pte-s5的数据。通过使用dtnb和抗ache损失方案两者在25℃下测定。
b在一些情况下,gd的两种s-对映异构体的解毒是双相的,这归因于两种毒性异构体spcr和spcs。在括号中给出慢阶段的参数。
c来自goldsmith等人[goldsmith,m.等人,arch.toxicol.,2016,90,pp.2711–2724.]的数据。使用监测ops的抗ache损失的方案,在37℃下用真正的神经试剂确定所有条目。
d来自goldsmith等人[goldsmith,m.和tawfik,d.s.,curr.opin.struct.biol.,2017,47,pp.140–150]的数据。
如表8中可见,相对于野生型pte,pte_28(seqidno:28)表现出vx水解效率的66倍增加,并且pte_29(seqidno:29)表现出水解rvx和gf的效率分别1,550倍和3,980倍的显著增加。
从pte_28(seqidno:28)开始,开始第二轮设计,这次指导funclib对第一轮中测试的最佳神经试剂水解酶中出现的3-5个突变的所有组合进行建模,并消除被预测为不稳定的(相对于pte_28(seqidno:28)的>8rosetta能量单位)的设计。对14种所得设计进行实验测试,发现设计pte_56(seqidno:56)和pte_57(seqidno:57)表现出对gd的活性增加(分别为32倍和122倍),并且两种设计均表现出水解gf的3,000倍增加。这些对高毒性神经试剂rvx、gd和gf具有kcat/km≥107m-1min-1的变体可能适用于体内解毒。
如从表8中进一步可见,通过测试63种变体观察到的效率增加与最佳变体相当,所述最佳变体来自应用十几轮的使用常规实验室进化策略的数千种变体的多样化和实验测试。此外,实验室进化实验要求对每种底物进行分开的选择活动,而设计的库包含数十种酶,其对我们测试的每种底物的效率提高。另外,所有变体都显示与高表达的dpte2(seqidno:1)起始序列相当的细菌表达水平(>300mg蛋白/升培养物)。
这些结果表明,pross和funclib的组合可能未表现出稳定性阈值瓶颈,所述瓶颈已限制许多酶(包括pte)的实验室进化。因此,funclib导致产生稳定且有效的酶的小、但功能上高度多样的库,并且在一些情况下可能绕过对高通量筛选的要求。
pte的序列空间:
表b呈现由本文呈现的方法(funclib)产生的氨基酸取代(突变)的序列空间,其利用上述关键残基并允许取代活性位点残基。所述序列空间具有8个氨基酸取代位置,各自在给定位置相比于wt(或起始序列)氨基酸具有至少一个任选取代,其中该位置的原始(野生型)氨基酸以粗体标记,并且是左起第一个。
实施例4
催化效率和选择性的结构基础
为了理解哪些分子因素是在通过实施本文提供的设计方法获得的一些变体中催化效率的高增加的基础,使用x-射线晶体学确定pte_6(seqidno:6)(对于2na,活性提高280倍)、pte_28(seqidno:28)(对于tbbl,活性提高65倍,且对于s-vx,活性提高103倍)和pte_29(seqidno:29)(对于gf,活性提高3980倍)的分子结构,并且结果呈现于图3和表9中。
图3呈现的图显示,根据本发明的一些实施方案,本文提供的pte变体中的设计突变表现出标志-上位性关系,其中每个圆圈代表dpte2(seqidno:1)的突变体,每个圆圈的面积与所述变体在水解芳酯乙酸2-萘酯(2na)中的比活性成正比,且其中pross设计和稳定的序列dpte2(seqidno:1)(其被用作本文提供的方法中的起点)表现出低比活性,并且每种点突变体表现出提高的比活性,比活性在双重突变体中下降,并且四重突变体设计pte_6(seqidno:6)相对于所有单一或双重突变体实质上提高比活性。
表9呈现pte设计的晶体学数据收集和精修统计,其中括号中的值是指相应高分辨率壳的数据。
结构见解:
晶体结构的视觉检查和位置分析揭示,相对于它们各自的模型,所有三种结构都显示高准确度(在骨架上均方根偏差[rmsd]<0.5å和在突变的活性位点残基中的0.3å全原子rmsd),证实设计过程导致产生如高效催化所要求的精确且预先组织的活性位点。
还将晶体结构与分子对接模拟中获得的结构进行比较,生成所述分子对接模拟以分别对pte_28(seqidno:28)、pte_29(seqidid:29)和pte_56(seqidno:56)的活性位点口袋中的vx、rvx和gd的毒性sp立体异构体建模。所得模型表明,设计的活动位点口袋足够大,足以容纳大体积的神经试剂并与其形成直接接触,这主要是由于两个大到小的取代his254gly和leu303thr(参见图3)。这些直接接触也可能是一些设计中观察到的高对映选择性(对于设计pte_29(seqidno:29),>104;参见表7)的基础。此外,几种改进的酯酶和内酯酶(pte_14-16(seqidno:14-16)、31-35(seqidno:31-35)和37(seqidno:37))编码his254arg突变,其改变活性位点口袋的空间和静电组织,如在实验室进化研究中报道的那样,其增强这些活性。因此得出结论,funclib-设计的突变主要影响活性位点口袋的结构,设计的库编码活性位点中的显著立体化学多样性,导致大的选择性变化,并且少数活性位点突变足以实现针对几种底物的催化效率和选择性的数量级提高。
设计的突变间的标志上位性:
根据本发明的一些实施方案,在pte的每种变体中,突变在空间上聚类。因此预期,一些设计将显示复杂的上位性关系,由此不能基于单点突变体的作用简单地预测多点突变体的作用。因此,测量了包含最佳设计中的三种的所有单点和双点突变体的比活性:pte_6(seqidno:6)、pte_28(seqidno:28)和pte_33(seqidno:33),其相对于pte分别具有四个、三个和四个活性位点突变(参见图4)。在pte_6(seqidno:6)和pte_33(seqidno:33)中,点突变相对于野生型提高催化效率,但一些双重突变体表现出的效率显著低于野生型的效率。
图4呈现作为根据本发明的一些实施方案在本文提供的pte变体中的选择性变化的基础的设计的活性位点口袋的立体化学特性的图示,其中pte_28(seqidno:28;在图4中表示为28)和pte_29(seqidno:29;在图4中表示为29)表现出比dpte2(seqidno:1;在图4中表示为1)更大的活性位点口袋以及针对大体积的v-和g-型神经试剂的高催化效率(从左上角起按顺时针顺序,分子渲染基于pdb条目:1hzy、6gbj、6gbk和6gbl;球体指示双金属中心的离子。
从图4中可见,pte_6(seqidno:6;在图4中表示为6)提供了令人信服的标志上位性情况,其中所有点突变都提高对于酯2na的比活性。然而,所有双突变体都比单点his257trp更差,并且三种双突变体甚至比起始点dpte2(seqidno:1;在图4中表示为1)更差。最显著的是,表现出比dpte2(seqidno:1;在图4中表示为1)更低的比活性的两种双突变体的组合(his254arg/his257trp和leu303thr/met317leu)导致产生活性最高的设计pte_6(seqidno:6;在图4中表示为6),其相对于dpte2(seqidno:1;在图4中表示为1)将比活性提高两个数量级,并且相对于leu303thr/met317leu双突变体将比活性提高三个数量级。此外,在dna水平上,点突变his→trp和leu→thr分别需要三个和两个核苷酸交换,通过逐步积累突变急剧降低pte_6(seqidno:6;在图4中表示为6)出现的几率。先前对导致临床分离的β-内酰胺酶突变体的适应性增强的突变轨迹的分析指出标志上位性在进化中无处不在;然而,在那种情况下,一部分轨迹显示单调且因此进化上可选择的活性的提高。相比之下,对于pte_6(seqidno:6;在图4中表示为6),目前呈现的分析表明甚至没有单调增加活性的单一突变轨迹。因此,本文提供的方法(funclib)可以接近无法通过逐步积累有益突变而获得的突变体,这是自然或实验室进化的前提条件。
尽管本发明已与其具体实施方案结合描述,但显而易见的是许多替代方案、修饰和变化对于本领域技术人员将是显而易见的。相应地,预期包含落入所附权利要求的精神和广泛范围内的所有此类替代方案、修饰和变化。
在本说明书中提及的所有出版物、专利和专利申请均通过引用以其整体并入本文说明书,其程度与每个个别出版物、专利或专利申请特别且个别指出通过引用并入本文相同。另外,在本申请中的任何参考文献的引用或鉴定不应解释为承认此类参考文献可作为本发明的现有技术。就使用部分标题的程度而言,它们不应解释为必要的限制。
另外,本申请的任何优先权文件在此以其整体通过引用并入本文。
- 还没有人留言评论。精彩留言会获得点赞!